

Case Report | DOI: https://doi.org/10.31579/2834-796X/005
Wilsons Disease presenting as Acute Pancreatitis with Non Gallstone Cholangitis: A Diagnostic Challenge
Associate Professor, Medicine, Ad-din Women’s Medical College Hospital, Dhaka, Bangladesh
*Corresponding Author: Richmond Ronald Gomes, Associate Professor of Medicine Ad-din Women’s Medical College Hospital, Dhaka, Bangladesh.
Citation: Richmond Ronald Gomes (2022). Wilsons Disease presenting as Acute Pancreatitis with Non Gallstone Cholangitis: A Diagnostic Challenge. International Journal of Cardiovascular Medicine, 1(1) DOI:10.31579/2834-796X/005
Copyright: © 2022 Richmond Ronald Gomes, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 22 September 2022 | Accepted: 30 September 2022 | Published: 05 October 2022
Keywords: wilsons;pancreatitis; cholangitis; amylase; lipase
Abstract
Wilson’s disease is a rare disorder of copper transport in hepatic cells, and may present as cholestatic liver disease; pancreatitis and cholangitis are rarely associated with Wilsons’s disease. In thepresent report, we describe a case of a 57-year-old man who was admitted with fever, jaundice and abdominal pain. The patient was diagnosed with acute pancreatitis with cholangitis based on raised serum amylase lipase and alkaline phosphatase level and ultrasonographically swollen pancreas. However, because of his long-term jaundice and hand tremor, the patient underwent further evaluation for Wilson’s disease, which was subsequently confirmed. This patient’s unique presentation exemplifies the overlap in the clinical and laboratory parameters of Wilson’s disease and cholestasis, and the difficulties associated with their differentiation. It suggests thatWilson’s disease should be considered in patients with pancreatitis, cholangitis, and protracted jaundice with neurological symptoms.
Introduction
Wilson’s disease is a rare, autosomal recessive disorder of copper transport in hepatic cells, with a reported incidence of 1: 30000 [1,2]. The disease may present as a cholestaticliver disease [1,2] or, rarely, as hemolytic anemia [3-8]. Mild pancreatitis, upon presentation, has been describedin only one case of Wilson’s disease and was attributed to copper deposition in the pancreas [9]. Cholelithiasis, asa result of hemolysis and pigmented gallstone formation, has also been reported in Wilson’s disease [10-15]. Theatypical clinical presentation of cholangitis in patients with normal findings on bile duct imaging has also beenreported16. However, pigmented gallstone pancreatitis and cholangitis, with concomitant obstructive jaundice,have not been reported as the presenting features of Wilson’s disease. The diagnosis of Wilson’s disease in the setting of obstructive jaundice and cholestatic disease can be challenging because the laboratory and liver biopsy results overlap with those of other cholestatic conditions. In the present report, we describe the case of a patient who presented with acute pancreatitis with cholangitis with normal bile duct, which was the first presenting feature of Wilson’s disease.
Case Report
A 57-year-old non-smoker man, not known to have diabetes, hypertension, coronary artery disease or bronchial asthma was admitted to our hospital with upper abdominal pain, vomiting and fever for 4 days. The pain was constant, gnawing and progressively increasing in intensity. There was radiation to back. Pain aggravated by taking meal and didn’t subside with medicines taken by him at home the name of which he could not mention. Along with pain he vomited for several times for the same duration. He also had high grade, intermittent fever with chills and rigor. Fever subsided temporarily after taking anti pyretic. He also stated that he was having yellow coloration of urine and sclera and hand tremor for the last few months. He reported no prior exposure to drugs, alcohol, or chemicals. None of his family member had liver disease. On admission, he was in good general condition, with a body temperature of 39.3°C, stable vital signs, and no distress His physical examination revealed bilateral scleral icterus and jaundiced skin as well as upper abdominal tenderness without hepatosplenomegaly, ascites or abdominal masses. Moreover, there were no signs of chronic liver disease, such as spider naevi, clubbing, gynaecomastia, engorged abdominal veins The results of the patient’s laboratory examination indicated a hemoglobin level of 10.2 g/dL (normal level: 14-18 g/dL); a leukocyte count of 14.6 k/μL (normal level: 4.0-11.0 k/μL); a platelet count of 252 k/μL (normal level: 150-450 k/ μL); a reticulocyte count of 1.1% (normal level: 0.5-1.5%); a serum amylase level of 462 U/L (normal level: 28-100 U/L); a serum lipase level of 376 U/L (normal level: 60-110 U/L serum SGPT 80 U/L (normal level: 0-35 U/L);. His results also demonstrated a serum alkaline phosphatase level of 317 U/L (normal level: 30-120 U/L), a total serum bilirubin level of 4.88 mg/dL (normal level: 0.3-1.2 mg/dL), a serum C-reactive protein level of 179 mg/L (normal level: 0-5 mg/L), an erythrocyte sedimentation rate of 76 mm/h (normal level: 0-15 mm/h), a serum albumin level of 3.36 g/dL (normal level: 3.5-5.2 g/dL). The patient’s creatinine level at admission was 0.94 mg/dL (normal level: 0.6-1.2 mg/dL). Fasting lipid profile, random blood sugar, serum electrolytes were normal on admission. But corrected serum calcium was low (8.2 mg/dl) and D-dimer was raised (3.58 mcg/ml, normal <0> yielded negative results. Tests for serum anti-nuclear also yielded negative results.
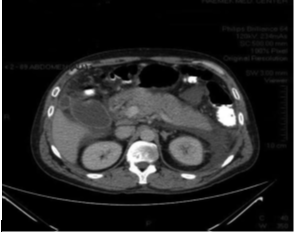
The patient was kept nothing per oral, Intravenous fluid, analgesic along with intravenous ciprofloxacin and metronidazole was started. With treatment he made significant clinical improvement. The presence of anemia and protracted jaundice led to the suspicion of Wilson’s disease, even though hemolytic episodes had not been observed. An ophthalmological slit-lamp examination failed to reveal Kayser-Fleischer rings. The rest of the physical examination, including a detailed neurological evaluation was unremarkable.On further investigation Serum ceruloplasmin 0.35g/dl U (normal 0.2-0.6 g/dl ). Spot Urinary copper was 278 mcg/dl (normal <60>
Discussion
Several metabolic disorders such as familial hyperlipoproteinemia or hypercalcemic states such as hyperparathyroidism may damage the exocrine pancreas and cause pancreatitis. Iron toxicosis in thalassemia and idiopathic hemochromatosis are other iron overload state affecting the pancreas. In such cases, the endocrine system is the main target, resulting in diabetes.
Wilson’s disease is an inherited disease, caused by one of several possible mutations in the ATP7B gene, leading to defective copper excretion into the bile and subsequent accumulation of copper in the liver, central nervous system, kidney, bone, blood, cornea, and endocrine glands, such as the parathyroid gland. The copper content of the pancreas in Wilson’s disease has only rarely been looked for. Neutron activation analysis of pancreatic tissue from a treated patient with Wilson’s disease at autopsy revealed elevated copper content, as compared with normal controls18.Glucose intolerance has been described19 in Wilson’s disease and may be attributed to damaged pancreatic beta cells due to copper deposition. Exocrine pancreatic function has been investi- gated in only a few studies. While a normal function has been described by Lankischet al.20, two other studies by Osswald Ct itl.21 and by Dreilingct al.22 have found abnormal pancreatic secretion in patients with Wilson’sdisease.
Excessive concentrations of copper are toxic to almost any component of the living cell—the plasma membrane, some proteins of the cytosol, and various organelles23. This may affect the cell membrane integrity, increase permeability, and cause Iysosomal breakage. Such effects may be responsible for the induction of pancreatitis when active proteolytic enzymes gain access to the pancreatic tissue, leading to auto digestion and inflammation. However, pancreatitis associated with Wilson’s disease has been described, to the best of our knowledge, in only two patients. Impaired venous drainage due to portal hyper tension was thought to be the likely cause of diffuse pancreatic congestion demonstrated ultrasonically in two patients. However, one of these patients had abdominal pain with very high amylase values, and subsequent penicillamine therapy resulted in abolition of hissymptoms [24]. In addition, another single patient has been described elsewhere. The pathogenesis of pancreatitis in the present case is supported by the penicillamine associated recovery and by exclusion of most other common causes of pancreatitis. The lack of fever, skin rash, or parotitis most probably excludes a viral cause such as mumps.
Pigment gallstones, a common finding inpatients with Wilson’s disease, may also induce pancreatitis. However, no evidence of this possibility has been demonstrated in the present case, since there was no evidence of active hemolysis and no appropriate ultrasonographic findings.
The hepatic manifestations of Wilson’s disease range from asymptomatic liver enzyme elevations to chronic, active hepatitis, cirrhosis, and acute fulminanthepatic failure [1,2]. Cholelithiasis has also been describedin patients with Wilson’s disease [10-15], including one study that found cholelithiasis among 25% of Wilson’s disease patients [20]. Cholelithiasis is frequently observed in female patients with Wilson’s disease, but its incidence is not high among male patients with the disease. Acute cholangitis with bile duct stones has not been described in Wilson’s disease, although edema of the gallbladder, mimicking acute cholecystitis and clinical cholangitis, but with normal bile duct imaging findings, has been reported [16,17].
The diagnosis of Wilson’s disease in the presence of cholestasis or long-term bile duct obstruction is difficult and challenging. Many characteristic laboratory findings in Wilson’s disease overlap with those of other cholestaticconditions. Cholestasis, due to conditions such as chronic active hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis, results in elevated 24-h urine copper levels. In such cases, the hepatic copper content is usually elevated, but the ceruloplasmin level may be either normal or elevate [25-29]. However, even in cases of severe cholestasis, the 24-h urine copper level does not usually exceed 200 μg/24 h2; in the present patient, the copper level was 300 μg/24 h. Liver biopsies in patients with Wilson’s disease yield non-specific findings and may indicate chronic, active, hepatitis-like results. The Kayser-Fleischer ring may also be absent in 50% of patients with hepatic Wilson’s disease.
The molecular analysis conducted on a blood sample from the present patient showed a homozygous V1140A peptide change (nucleotide change, c.3419 T>C) mutation in the ATP7B gene. Over 500 mutations of the ATP7B gene have been reported to cause Wilson’s disease [2,30], some of which are very rare. These mutations lead to the production of defective copper transporter proteins, resulting in copper accumulation in the tissue. The V1140A mutation is a missense mutation, or sequence polymorphism in codon 3419 T > C of exon 16 on chromosome 13q, which was previously described in a Chinese, Czech-Slovakian, and Thai siblings and in aYugoslavian cohort with Wilson’s disease [31-33].
A single, specific test for diagnosing Wilson’s disease does not exist. Therefore, the European Association forthe Study of the Liver practical guidelines established a Wilson’s disease scoring system25. The diagnostic score is based on six available tests, including the presence of the Kayser-Fleischer ring, the presence of neurologic symptoms, ceruloplasmin values, and the presence of hemolytic anemia, liver copper content in the absence of cholestasis, urinary copper levels, and a mutation analysis. A score of greater than or equal to four establishes a diagnosis of Wilson’s disease25,34; the present patient had a score of six, clearly indicating a diagnosis of Wilson’s disease. The low serum uric acid level found in our patient may be characteristic, although not diagnostic, of Wilson’s disease and is caused by renal tubular defects2. The reversible coagulopathy with anemia observed in the present patient has also been previously described in a patient with Wilson’s disease16. The pigmented gallstones associated with Wilson’s disease are related to hemolytic episodes and might be a presenting feature of the disease2-8. In the present patient, the reticulocyte production index was higher than normal whereas the serum haptoglobin level, which is usually decreased in patients with hemolysis, was within normal limits. We hypothesize that this may be due to the presence of acute pancreatitis and cholangitis, considering the fact that haptoglobin is characterized as an acute phase reactant.
Conclusion
Since gastrointestinal symptoms such as abdominal pain and nausea are quite common in Wilson’s disease, we suggest that the possibility of pancreatitis and cholangitis should be considered in untreated patients and in patients who have protracted cholestasis and an obscure disease course
References
- Ferenci P. Review article: diagnosis and current therapy ofWilson’s disease. Aliment PharmacolTher 2004; 19: 157-165
View at Publisher | View at Google Scholar - Roberts EA,Schilsky ML. Diagnosis and treatment of Wilsondisease: an update. Hepatology2008; 47: 2089-2111
View at Publisher | View at Google Scholar - Członkowska A, Gromadzka G, Büttner J, Chabik G. Clinicalfeatures of hemolysis, elevated liver enzymes, and lowplatelet count syndrome in undiagnosed Wilson disease:report of two cases. Arch GynecolObstet2010; 281: 129-134[PMID: 19381668 DOI: 10.1007/s00404-009-1080-6]
View at Publisher | View at Google Scholar - Aagaard NK, Thomsen KL, Holland-Fischer P, JørgensenSP, Ott P. A 15-year-old girl with severe hemolytic Wilson’scrisis recovered without transplantation after extracorporealcirculation with the Prometheus system. Blood Purif2009; 28:102-107 [PMID:19439930 DOI: 10.1159/000218090]
View at Publisher | View at Google Scholar - Liapis K, Charitaki E, Delimpasi S. Hemolysis in Wilson’s disease. Ann Hematol2011; 90: 477-478 [PMID: 20683594DOI: 10.1007/s00277-010-1038-6]
View at Publisher | View at Google Scholar - Balkema S, Hamaker ME, Visser HP, Heine GD, Beuers U.Haemolyticanaemia as a first sign of Wilson’s disease. NethJ Med 2008; 66: 344-347 [PMID: 18809982]
View at Publisher | View at Google Scholar - Maple JT, Litin SC. 26-Year-old man with rapidly progressivejaundice and anemia. Mayo ClinProc 2002; 77: 83-86[PMID: 11794461]
View at Publisher | View at Google Scholar - El Khattabi A,Seddik H, Fatihi J, Salaheddine H, BadaouiM, Amézyane T, Mahassine F, Ohayon V. [Acute recurrenthemolytic anemia as the first manifestation of Wilson’s disease: Report of a case]. TransfusClinBiol2009; 16: 39-42[PMID: 19329346 DOI: 10.1016/j.tracli.2009.01.005]
View at Publisher | View at Google Scholar - Weizman Z, Picard E, Barki Y, Moses S. Wilson’s disease associatedwith pancreatitis. J Pediatr GastroenterolNutr1988; 7:931-933 [PMID: 3199280]
View at Publisher | View at Google Scholar - Akhan O, Akpinar E, Oto A, Köroglu M, Ozmen MN, AkataD, Bijan B. Unusual imaging findings in Wilson’s disease.EurRadiol2002; 12Suppl 3: S66-S69 [PMID: 12522607]
View at Publisher | View at Google Scholar - Dupuy R, Vallin J, Fabiani F. [2 cases of Wilson’s diseasewith hepatic precession in biliary lithiasis]. Rev IntHepatol1969; 19: 233-240 [PMID: 5404531]
View at Publisher | View at Google Scholar - Rosenfield N, Grand RJ, Watkins JB, Ballantine TV, LeveyRH. Cholelithiasis and Wilson disease. J Pediatr1978; 92:210-213 [PMID: 621603]
View at Publisher | View at Google Scholar - Walshe JM. Wilson’s disease: gall stone copper followingliver transplantation. Ann ClinBiochem 1998; 35(Pt 5):681-682 [PMID: 9768338]
View at Publisher | View at Google Scholar - Singh R, Sibal A, Jain SK. Gall stones, G-6PD deficiencyand Wilson’s disease. Indian J Pediatr2002; 69: 635 [PMID:12173707]
View at Publisher | View at Google Scholar - Ghosh JB, Chakrabarty S, Singh AK, Gupta D. Wilson’sdisease--unusual features. Indian J Pediatr 2004; 71: 937-938[PMID: 15531840]
View at Publisher | View at Google Scholar - Wadera S, Magid MS, McOmber M, Carpentieri D, Miloh T. Atypical presentation of Wilson disease. Semin Liver Dis 2011; 31: 319-326 [PMID: 21901661 DOI: 10.1055/s-0031-1286062]
View at Publisher | View at Google Scholar - Chang SK, Chan CL, Yu RQ, Wai CT. Mimicry of acute cholecystitis from Wilson’s disease. Singapore Med J 2009; 50: e102-e104 [PMID: 19352551]
View at Publisher | View at Google Scholar - Len ML. Strickland GT, Yeh SJ. Tissue copper, zinc, and manganese levels in Wilson’x disease: studies with the use of neutron activation analysis. J LubClinMeJ1971;77:43M4.
View at Publisher | View at Google Scholar - Iohansen K. GregersenG . Glucose intolerance in W ilson’s disease. Ari’li Intern Alert 1972; 129:557—590.
View at Publisher | View at Google Scholar - LankischPG ,Kaboth U . Koop H. PankreasbeteiligungbeiMorbusWilson'?
View at Publisher | View at Google Scholar - Osswald P, N ieseen K H .Exokrine Pankreasinsuffixienz beMorbusWi1son.KlinW'rx/ieri,i‹/ir1976;54:539—543.
View at Publisher | View at Google Scholar - Dreiling DA, GrateronH .Studies in pancreatic secretionV111. Pancreatic function in patients with Wilson’ sdisease198.3;50:335—337.
View at Publisher | View at Google Scholar - Sternlie b 1. Copper and the liver. Giustr‹›ent‹•rologi’ 1980;7S:161fi—25.
View at Publisher | View at Google Scholar - MinnsRA,EdenOB.HendryGMA.Pancreaticenlargementand Wilson’s disease [Letter). Lnrir ci 1982:2:337—338.
View at Publisher | View at Google Scholar - European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol2012; 56: 671-685 [PMID: 22340672 DOI: 10.1016/j.jhep.2011.11.007]
View at Publisher | View at Google Scholar - Smallwood RA, Williams HA, Rosenoer VM, Sherlock S. Liver-copper levels in liver disease: studies using neutron activation analysis. Lancet 1968; 2: 1310-1313 [PMID:4177386]
View at Publisher | View at Google Scholar - Ritland S, Steinnes E, Skrede S. Hepatic copper content, urinary copper excretion, and serum ceruloplasmin in liver disease. Scand J Gastroenterol1977; 12: 81-88 [PMID: 834974]
View at Publisher | View at Google Scholar - Benson GD. Hepatic copper accumulation in primary biliary cirrhosis. Yale J Biol Med 1979; 52: 83-88 [PMID: 452626]
View at Publisher | View at Google Scholar - Gross JB, Ludwig J, Wiesner RH, McCall JT, LaRusso NF. Abnormalities in tests of copper metabolism in primary sclerosing cholangitis. Gastroenterology 1985; 89: 272-278 [PMID: 4007418]
View at Publisher | View at Google Scholar - Rosencrantz R, SchilskyM. Wilson disease: pathogenesis and clinical considerations in diagnosis and treatment. Semin Liver Dis 2011; 31: 245-259 [PMID: 21901655 DOI: 10.1055/s-0031-1286056]
View at Publisher | View at Google Scholar - Liu XQ, Zhang YF, Liu TT, Hsiao KJ, Zhang JM, Gu XF, BaoKR, Yu LH, Wang MX. Correlation of ATP7B genotype withphenotype in Chinese patients with Wilson disease. World JGastroenterol 2004; 10: 590-593 [PMID: 14966923]
View at Publisher | View at Google Scholar - Gojová L, Jansová E, Külm M,Pouchlá S, Kozák L. Genotyping microarray as a novel approach for the detection of ATP7B gene mutations in patients with Wilson disease. Clin Genet 2008; 73: 441-452 [PMID: 18371106 DOI: 10.1111/ j.1399-0004.2008.00989.x]
View at Publisher | View at Google Scholar - Treepongkaruna S, Pienvichit P, Phuapradit P, KodcharinP, Wattanasirichaigoon D. Mutations of ATP7B gene in two Thai siblings with Wilson disease. Asian Biomed 2010; 4: 163-169
View at Publisher | View at Google Scholar - Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, Schilsky M, Cox D, Berr F. Diagnosis and phenotypicclassification of Wilson disease. Liver Int2003; 23: 139-142 [PMID: 12955875]
View at Publisher | View at Google Scholar -
View at Publisher | View at Google Scholar -
View at Publisher | View at Google Scholar -
View at Publisher | View at Google Scholar
