

Review Article | DOI: https://doi.org/10.31579/2834-8508/026
The Role of Genetic Mutations on Gene FOXL2 in Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome
- Maryam Montazeri
- Shiva Ghayor
- Shahin Asadi *
Division of Medical Genetics and Molecular Pathology Research, Harvard University, Boston Children's Hospital,USA.
*Corresponding Author: Shahin Asadi, Division of Medical Genetics and Molecular Pathology Research, Harvard University, Boston Children's Hospital, USA.
Citation: Maryam Montazeri, Shiva Ghayor, Shahin Asadi. (2024). The Role of Genetic Mutations on Gene FOXL2 in Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome. Archives of Clinical and Experimental Pathology. 3(2); Doi:10.31579/2834-8508/026
Copyright: © 2024 Shahin Asadi, this is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 15 February 2024 | Accepted: 28 February 2024 | Published: 06 March 2024
Keywords: BPEI Syndrome, Genetic Mutation, FOXL2 Gene, Infertility
Abstract
BPEI syndrome is a genetic disorder that mainly affects the development of the eyelids. People with this disease have blepharophimosis, drooping eyelids (ptosis) and abnormal skin on top of the eyelids near the inner corner of the eye (epicanthus inversus). In addition to the above, other structures in the eyes and face may also be affected by BPEI syndrome. BPEI syndrome types 1 and 2 are caused by the mutation of the FOXL2 gene, which is located in the long arm of chromosome 3 as 3q22.3.
Overview of BPEI Syndrome
BPEI syndrome is a genetic disorder that mainly affects the development of the eyelids. People with this disease have blepharophimosis, drooping eyelids (ptosis) and abnormal skin on top of the eyelids near the inner corner of the eye (epicanthus inversus). In addition, the distance between the eyes (hypertelorism) is also present in BPEI patients. Because of these eyelid abnormalities, the eyelids cannot open fully, and vision may be limited.[1]
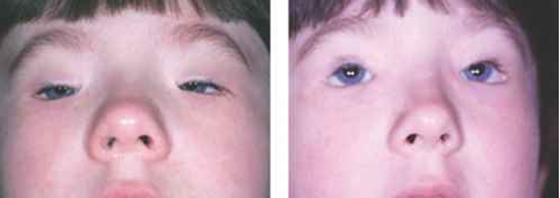
Figure 1: Image of children with BPEI syndrome with distinctive facial features.1
Clinical Signs and Symptoms of BPEI Syndrome
In addition to the above, other structures in the eyes and face may also be affected by BPEI syndrome. People affected by BPEI syndrome are at risk of developing vision problems such as nearsightedness (myopia) or farsightedness (hyperopia) that begin in childhood. They may have strabismus or lazy eye (amblyopia) that affects one or both eyes. People with BPEI syndrome may have distinctive facial features such as a wide nose bridge, low-set ears, or a short distance between the upper lip and nose (short philtrum).[1,2]
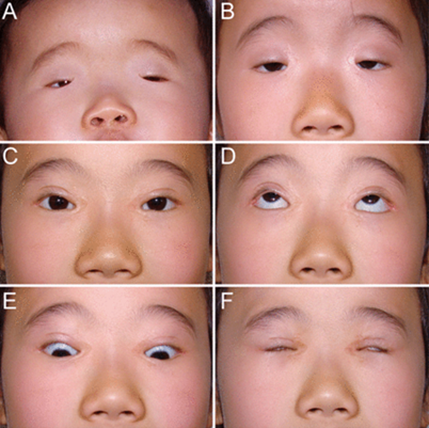
Figure 2: Image of eye disorders in BPEI patients.[1]
There are two types of BPEI syndrome, each characterized by its own signs and symptoms. Eyelid abnormalities and other facial features are common to both types 1 and 2 of BPEI syndrome. BPEI type 1 syndrome is also associated with early or premature ovarian failure in women, which causes them to have shorter menstrual periods and eventually stop menstruating before the age of 40. Primary ovarian failure can cause miscarriage or complete inability to conceive (infertility).[1,2]
Etiology of BPEI Syndrome
BPEI syndrome types 1 and 2 are caused by the mutation of the FOXL2 gene, which is located in the long arm of chromosome 3 as 3q22.3. This gene provides the necessary instructions for the synthesis of a protein that is active in the eyelids and ovaries. FOXL2 protein is probably involved in the development of eyelid muscles. Before birth and in adulthood, this protein regulates the growth and development of specific ovarian cells and the breakdown of specific molecules.[1,3]
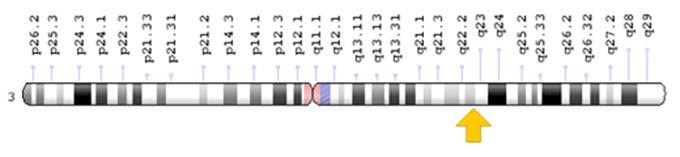
Figure 3: Schematic of chromosome number 3, where the FOXL2 gene is located in the long arm of this chromosome as 3q22.3.1
It is difficult to predict the type of BPEI syndrome that results from multiple mutations of the FOXL2 gene. However, mutations that result in partial loss of FOXL2 function generally cause BPEI type II syndrome. These mutations probably disrupt the regulation of normal muscle growth in the eyelids, resulting in incomplete eyelids that cannot open fully. Mutations that result in complete loss of function of the FOXL2 protein often cause BPEI type I syndrome. These mutations disrupt the regulation of eyelid growth as well as various activities in the ovaries, which leads to abnormality of the eyelid and abnormal maturation, and the breakdown of some ovarian cells and premature death of the egg cells.[1,4]
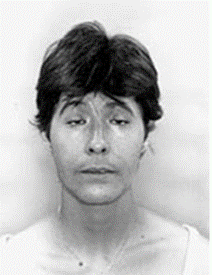
Figure 4: Another view of eye disorders including ptosis in a person with BPEI syndrome.[1]
BPEI syndrome follows an autosomal dominant inheritance pattern. Therefore, to create this syndrome, a copy of the mutated FOXL2 gene (from both parents) is needed, and the chance of having a child with this syndrome in an autosomal dominant state is 50% for each possible pregnancy.[1,4]
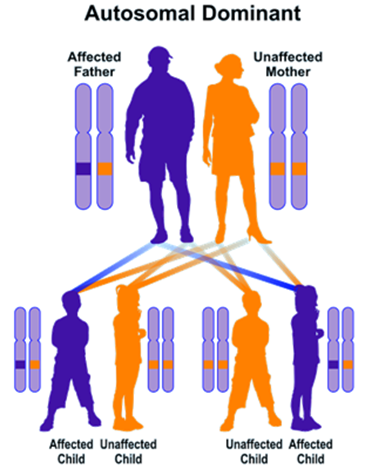
Figure 5: Schematic view of the autosomal dominant inheritance pattern that BPEI syndrome also follows.[1]
Frequency of BPEI Syndrome
BPEI syndrome is a genetic disorder whose frequency is not known in the world.[1,5]
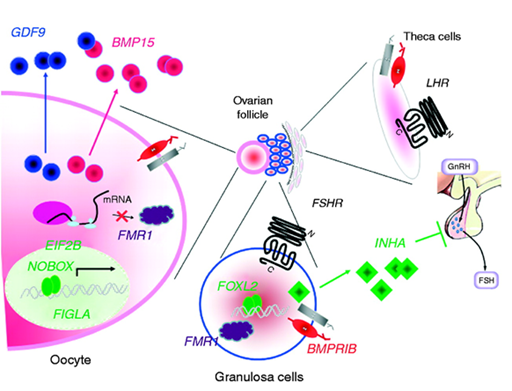
Figure 6: Schematic of the molecular pathway of the FOXL2 gene in ovarian cells.[1]
Diagnosis of BPEI Syndrome
BPEI syndrome is diagnosed based on clinical and physical findings of patients and some pathological tests. The most accurate method of diagnosing this syndrome is molecular genetic testing for the FOXL2 gene in order to check the presence of possible mutations[1,6].
Treatment options for BPEI Syndrome
The treatment and management strategy of BPEI syndrome is symptomatic and supportive. Treatment may be done with the efforts and coordination of a team of specialists, including ophthalmologists, otolaryngologists, surgeons, and other health care professionals. There is no definitive treatment for this syndrome and all clinical measures are aimed at alleviating the suffering of the sufferers. Genetic counseling is also necessary for all parents who want a healthy child.[1,7]
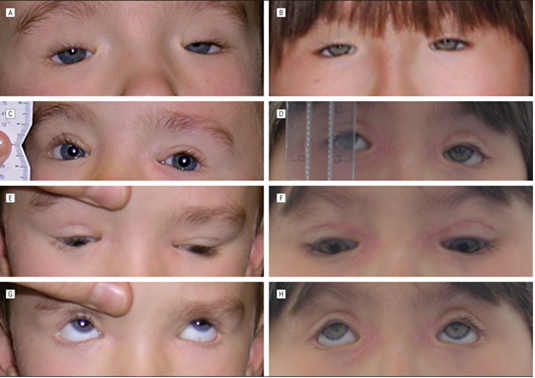
Figure 7: Images of related disorders in BPEI patients.[1]
Discussion and Conclusion
People affected by BPEI syndrome are at risk of developing vision problems such as nearsightedness (myopia) or farsightedness (hyperopia) that begin in childhood. They may have strabismus or lazy eye (amblyopia) that affects one or both eyes. It is difficult to predict the type of BPEI syndrome that results from multiple mutations of the FOXL2 gene. However, mutations that result in partial loss of FOXL2 function generally cause BPEI type II syndrome. Mutations that result in complete loss of function of the FOXL2 protein often cause BPEI type I syndrome. The treatment and management strategy of BPEI syndrome is symptomatic and supportive. Treatment may be done with the efforts and coordination of a team of specialists, including ophthalmologists, otolaryngologists, surgeons, and other health care professionals.[1,8]
References
- Asadi S, The Book of Human Genetic Infertility & Sterility, Amidi Publications, Iran 2020.
View at Publisher | View at Google Scholar - Beysen D, De Jaegere S, Amor D, Bouchard P, Christin-Maitre S, Fellous M, Touraine P, Grix AW, Hennekam R, Meire F, Oyen N, Wilson LC, Barel D, Clayton-Smith J, de Ravel T, Decock C, Delbeke P, Ensenauer R, Ebinger F, Gillessen-Kaesbach G, Hendriks Y, Kimonis V, Laframboise R, Laissue P, Leppig K, Leroy BP, Miller DT, Mowat D, Neumann L, Plomp A, Van Regemorter N, Wieczorek D, Veitia RA, De Paepe A, De Baere E. Identification of 34 novel and 56 known FOXL2 mutations in patients with Blepharophimosis syndrome. Hum Mutat. 2008 Nov;29(11):E205-19. doi: 10.1002/humu.20819.
View at Publisher | View at Google Scholar - Beysen D, De Paepe A, De Baere E. FOXL2 mutations and genomic rearrangements in BPES. Hum Mutat. 2009 Feb;30(2):158-69. doi: 10.1002/humu.20807. Review.
View at Publisher | View at Google Scholar - Beysen D, Raes J, Leroy BP, Lucassen A, Yates JR, Clayton-Smith J, Ilyina H, Brooks SS, Christin-Maitre S, Fellous M, Fryns JP, Kim JR, Lapunzina P, Lemyre E, Meire F, Messiaen LM, Oley C, Splitt M, Thomson J, Van de Peer Y, Veitia RA, De Paepe A, De Baere E. Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am J Hum Genet. 2005 Aug;77(2):205-18. Epub 2005 Jun 16.
View at Publisher | View at Google Scholar - Choi KH, Kyung S, Oh SY. The factors influencing visual development in blepharophimosis-ptosis-epicanthus inversus syndrome. J Pediatr Ophthalmol Strabismus. 2006 Sep-Oct;43(5):285-8.
View at Publisher | View at Google Scholar - D'haene B, Nevado J, Pugeat M, Pierquin G, Lowry RB, Reardon W, Delicado A, García-Miñaur S, Palomares M, Courtens W, Stefanova M, Wallace S, Watkins W, Shelling AN, Wieczorek D, Veitia RA, De Paepe A, Lapunzina P, De Baere E. FOXL2 copy number changes in the molecular pathogenesis of BPES: unique cohort of 17 deletions. Hum Mutat. 2010 May;31(5):E1332-47. doi: 10.1002/humu.21233.
View at Publisher | View at Google Scholar - Dipietromaria A, Benayoun BA, Todeschini AL, Rivals I, Bazin C, Veitia RA. Towards a functional classification of pathogenic FOXL2 mutations using transactivation reporter systems. Hum Mol Genet. 2009 Sep 1;18(17):3324-33. doi: 10.1093/hmg/ddp273. Epub 2009 Jun 10.
View at Publisher | View at Google Scholar - Taylor A, Strike PW, Tyers AG. Blepharophimosis-ptosis-epicanthus inversus syndrome: objective analysis of surgical outcome in patients from a single unit. Clin Experiment Ophthalmol. 2007 Apr;35(3):262-9.
View at Publisher | View at Google Scholar
