

Review Article | DOI: https://doi.org/10.31579/2834-8664/001
Sustained Release Floating Matrix Tablets of Ciprofloxacin Using Combination of Different Polymers
- Divakar Kanakagiri 1
Head of Operations at Joulepoint Pvt. Ltd Hyderabad, Telangana, India.
*Corresponding Author: Divakar Kanakagiri, Head of Operations at Joulepoint Pvt. Ltd Hyderabad, Telangana, India
Citation: Divakar Kanakagiri, (2022) Sustained Release Floating Matrix Tablets of Ciprofloxacin Using Combination of Different Polymers. International Journal of clinical and Medical Case Reports, 1(1); Doi: 10.31579/2834-8664/01
Copyright: © 2022 Divakar Kanakagiri, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 08 September 2022 | Accepted: 15 September 2022 | Published: 23 September 2022
Keywords: cross-over trial; background therapy; dosing discontinuation; observational study; health authority
Abstract
Drug delivery to the exact site of action become the key to the overall effectiveness of the drug, but when it comes to gastric ulcers it is a rate determining parameter and most crucial factor to play in the treatment of ulcers.More often, drug absorption is unsatisfactory and highly variable among and between individuals, despite excellent in vitro release patterns. The reasons for this are essentially physiological and usually affected by the GI transit of the form, especially its gastric residence time (GRT), which appears to be one of the causes of the overall transit time variability. Over the past three decades, the pursuit and exploration of devices designed to be retained in the upper part of the gastrointestinal (GI) tract has advanced consistently in terms of technology and diversity, encompassing a variety of systems and devices such as floating systems, raft systems, expanding systems, swelling systems, bioadhesive systems and low-density systems. Gastric retention will provide advantages such as the delivery of drugs with narrow absorption windows in the small intestinal region. Also, longer residence time in the stomach could be advantageous for local action in the upper part of the small intestine, for example treatment of peptic ulcer disease .The present research work aims to formulate and evaluate sustained release floating matrix tablets of Ciprofloxacin using combination of different polymers.
Introduction
Historically, oral drug administration has been the predominant route for drug delivery. During the past two decades, numerous oral delivery systems have been developed to act as drug reservoirs from which the active substance can be released over a defined period of time at a predetermined and controlled rate. From a pharmacokinetic point of view, the ideal sustained and controlled release dosage form should be comparable with an infusion, which supplies continuously the amount of drug needed to maintain constant plasma levels once the steady state is reached [1].
Although some important applications, including oral administration of peptide and protein drugs, can be used to prepare colonic drug delivery systems, targeting drugs to the colon by the oral route. More often, drug absorption is unsatisfactory and highly variable among and between individuals, despite excellent in vitro release patterns. The reasons for this are essentially physiological and usually affected by the GI transit of the form, especially its gastric residence time (GRT), which appears to be one of the causes of the overall transit time variability [2].
Over the past three decades, the pursuit and exploration of devices designed to be retained in the upper part of the gastrointestinal (GI) tract has advanced consistently in terms of technology and diversity, encompassing a variety of systems and devices such as floating systems, raft systems, expanding systems, swelling systems, bioadhesive systems and low-density systems. Gastric retention will provide advantages such as the delivery of drugs with narrow absorption windows in the small intestinal region. Also, longer residence time in the stomach could be advantageous for local action in the upper part of the small intestine, for example treatment of peptic ulcer disease.
Furthermore, improved bioavailability is expected for drugs that are absorbed readily upon release in the GI tract. These can be delivered ideally by slow release from the stomach. Many drugs categorised as once-a-day delivery have been demonstrated to have suboptimal absorption due to dependence on the transit time of the dosage form, making traditional extended release development challenging. Therefore, a system designed for longer gastric retention will extend the time within which drug absorption can occur in small intestine [3].
Certain types of drugs can benefit from using gastric retentive
devices. These include:
•Acting locally in the stomach.
• Primarily absorbed in the stomach.
• Poorly soluble at an alkaline pH.
• Narrow window of absorption.
• Absorbed rapidly from the GI tract.
• Degrade in the colon.
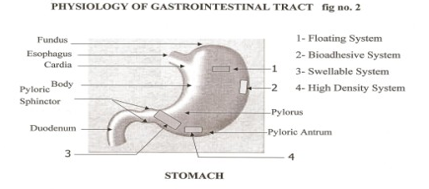
- Physiology Of The Stomach
The Gastrointestinal is a tube about nine metres long that runs through the middle of the body from the mouth to the anus and includes the throat (pharynx), oesophagus, stomach, small intestine (consisting of the duodenum, jejunum and ileum) and large intestine
(consisting of the cecum, appendix, colon and rectum). The wall of the Gatrointestinal tract has the same general structure throughout most of its length from the oesophagus to the anus, with some local variations for each region. The stomach is an organ with a capacity for storage and mixing. The antrum region is responsible for the mixing and grinding of gastric contents.
Under fasting conditions, the stomach is a collapsed bag with a residual volume of approximately 50ml and contains a small amount of gastric fluid (pH 1–3) and air. The mucus spreads and covers the mucosal surface of the stomach as well as the rest of the GI tract. The GI tract is in a state of continuous motility consisting of two modes, interdigestive motility pattern and digestive motility pattern. The former is dominant in the fasted state with a primary function of cleaning up the residual content of the upper GI tract. The interdigestive motility pattern is commonly called the ‘migrating motor complex’ (‘MMC’) and is organized in cycles of activity and quiescence [4].
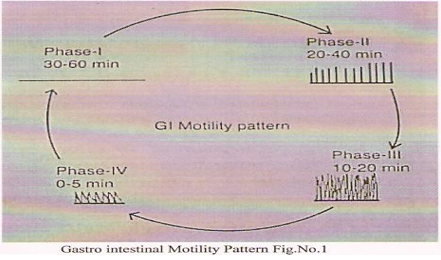
Each cycle lasts 90–120 minutes and consists of four phases. The concentration of the hormone motilin in the blood controls the duration of the phases. In the interdigestive or fasted state, an MMC wave migrates from the stomach down the GI tract every 90–120 minutes. A full cycle consists of four phases, beginning in the lower oesophageal sphincter/ gastric pacemaker, propagating over the whole stomach, the duodenum and jejunum, and finishing at the ileum. Phase III is termed the ‘housekeeper wave’ as the powerful contractions in this phase tend to empty the Stomach of its fasting contents and indigestible debris. The administration and subsequent ingestion of food rapidly interrupts the MMC cycle, and the digestive phase is allowed to take place. The upper part of the stomach stores the ingested food initially, where it is compressed gradually by the phasic contractions.
The digestive or fed state is observed in response to meal ingestion. It resembles the fasting Phase II and is not cyclical, but continuous, provided that the food remains in the stomach. Large objects are retained by the stomach during the fed pattern but are allowed to pass during Phase III of the interdigestive MMC. It is thought that the sieving efficiency (i.e. the ability of the stomach to grind the food into smaller size) of the stomach is enhanced by the fed pattern or by the presence of food [5].
The fasted-state emptying pattern is independent of the presence of any indigestible solids in the stomach. Patterns of contractions in the stomach occur such that solid food is reduced to particles of less than 1mm diameter that are emptied through the pylorus as a suspension. The duration of the contractions is dependent on the physiochemical characteristics of the ingested meal [6].
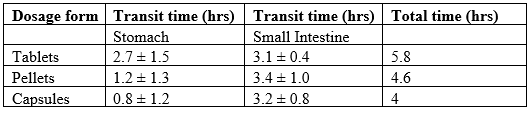
Generally, a meal of ~450kcal will interrupt the fasted state motility for about three to four hours. It is reported that the antral contractions reduce the size of food particles to ≤1mm and propel the food through the pylorus. However, it has been shown that ingestible solids ≤7mm can empty from the fed stomach in humans. TableNo.1

P – Passive diffusion C – Aqueous channel transport
A – Active transport F – Facilitated transport
I – Ion-pair transport E – Entero-or pinocytosis
CM – Carrier mediated transport
1.1.1 Different Features Of Stomach
Gastric pH: Fasted healthy subject 1.1 ± 0.15
Fed healthy subject 3.6 ± 0.4
Volume : Resting volume is about 25-50 ml
Gastric secretion: Acid, pepsin, gastrin, mucus and some enzymes about 60 ml with approximately 4 mmol of hydrogen ions per hour.
Effect of food on Gastric secretion: About 3 liters of secretions are added to the food. Gastro intestinal transit time Figure No.1
Table 1: Salient Features of Upper Gastrointestinal Tract
1.1.2 Requirements For Gastric Retention:
Physiological factors in the stomach, it must be noted that, to achieve gastric retention, the dosage form must satisfy certain requirements. One of the key issues is that the dosage form must be able to withstand the forces caused by peristaltic waves in the stomach and the constant contractions and grinding and churning mechanisms. To function as a gastric retention device, it must resist premature gastric emptying. Furthermore, once its purpose has been served, the device should be removed from the stomach with ease.
NEED FOR GASTRO RETENTION:7
- Drugs that are absorbed from the proximal part of the gastrointestinal tract (GIT).
- Drugs that are less soluble or are degraded by the alkaline pH they encounters at the lower part of GIT.
- Drugs that are absorbed due to variable gastric emptying time.
- Local or sustained drug delivery to the stomach and proximal Small intestine to treat
- certain conditions.
- Particularly useful for the treatment of peptic ulcers caused by H. Pylori Infections.
1.2.1 Advantages of Gastroretentive Delivery Systems:8
- Improvement of bioavailability and therapeutic efficacy of the drugs and possible reduction of dose e.g. Furosemide
- Maintenance of constant therapeutic levels over a prolonged period and thus reduction in fluctuation in therapeutic levels minimizing the risk of resistance especially in case of antibiotics. e.g. b-lactam antibiotics (penicillins and cephalosporins)
- Retention of drug delivery systems in the stomach prolongs overall.
- Gastrointestinal transit time thereby increasing bioavailability of sustained release delivery systems intended for once-a-day administration. e.g. Ofloxacin
1.2.2 Limitations of the techniques of Gastro retention:
- More predictable and reproducible floating properties should be achieved in all the extreme gastric conditions.
- The floating systems in patients with achlorhydria can be questionable in case of swellable systems, faster swelling properties are required and complete swelling of the system should be achieved well before the gastric emptying time.
- Bioadhesion in the acidic environment and high turnover of mucus may raise questions about the effectiveness of this technique. Similarly retention of high density systems in the antrum part under the migrating waves of the stomach is questionable.
- Not suitable for drugs that may cause gastric lesions e.g. Non- steroidal anti inflammatory drugs. Drugs that are unstable in the strong acidic environment, these systems do not offer significant advantages over the conventional dosage forms for drugs that are absorbed throughout the gastrointestinal tract.
- The mucus on the walls of the stomach is in a state of constant renewal, resulting in unpredictable adherence.
- In all the above systems the physical integrity of the system is very important and primary requirement for the success of these systems.
1.2.3 Factors Affecting Gastric Retention:
1.2.3.1 Particle size and pH of the stomach
Gastric residence time of an oral dosage form is affected by several factors. To pass through the pyloric valve into the small intestine the particle size should be in the range of 1 to 2 mm. The pH of the stomach in fasting state is ~1.5 to 2.0 and in fed state is 2.0 to 6.0. A large volume of water administered with an oral dosage form raises the pH of stomach contents to 6.0 to 9.0. Stomach doesn’t get time to produce sufficient acid when the liquid empties the stomach, hence generally basic drugs have a better chance of dissolving in fed state than in a fasting state.
1.2.3.2 Concomitant
Fassihi and Yang (1998) developed a zero-order controlled release multilayer tablet composed of at least 2 barrier layers and 1 drug layer. All the layers were made of swellable, erodible polymers and ingestion of food ant its nature, caloric content and frequency of intake.
The rate of gastric emptying depends mainly on viscosity, volume, and caloric content of meals. Nutritive density of meals helps determine gastric emptying time. It does not make any difference whether the meal has high protein, fat, or carbohydrate content as long as the caloric content is the same. However, increase in acidity and caloric value slows down gastric emptying time.
1.2.3.3 Biological factors.
Biological factors such as age, body mass index (BMI), gender, posture, and diseased states (diabetes, Chron’s disease) influence gastric emptying. In the case of elderly persons, gastric emptying is slowed down. Generally females have slower gastric emptying rates than males. Stress increases gastric emptying rates while depression slows it down. The resting volume of the stomach is 25 to 50 ml. Volume of liquids administered affects the gastric emptying time.
When volume is large, the emptying is faster. Fluids taken at body temperature leave the stomach faster than colder or warmer fluids. Studies have revealed that gastric emptying of a dosage form in the fed state can also be influenced by its size. Small-size tablets leave the stomach during the digestive phase while the large-size tablets are emptied during the housekeeping waves. It has been demonstrated using radio labeled technique that there is a difference between gastric emptying times of a liquid, digestible solid, and indigestible solid. It was suggested that the emptying of large (>1 mm) indigestible objects from stomach was dependent upon interdigestive migrating myoelectric complex.
When liquid and digestible solids are present in the stomach, it contracts ~3 to 4 times per minute leading to the movement of the contents through partially opened pylorus. Indigestible solids larger than the pyloric opening are propelled back and several phases of myoelectric activity take place when the pyloric opening increases in size during the housekeeping wave and allows the sweeping of the indigestible solids. Studies have shown that the gastric residence time (GRT) can be significantly increased under the fed conditions since the MMC is delayed.
Several formulation parameters can affect the gastric residence time. More reliable gastric
emptying patterns are observed for multiparticulate formulations as compared with single unit
formulations, which suffer from “all or none concept.” gastrointestinal tract, their transport is
affected to a lesser extent by the transit time of food compared with single unit formulation.
1.2.3.4 Size and Shape of the Device:
Timmermans and Andre, 1989 studied the effect of size of floating and non-floating dosage forms on gastric emptying and concluded that the floating units remained buoyant on gastric fluids. These are less likely to be expelled from the stomach compared with the non-floating units, which lie in the antrum region and are propelled by the peristaltic movement
Table 2: Transit times of various dosage forms across the segments of the GIT.
Size and shape of dosage unit also affect the gastric emptying. Garg and Sharma reported that tetrahedron- and ring-shaped devices have a better gastric residence time as compared with other shapes. The diameter of the dosage unit is also equally important as a formulation parameter. Dosage forms having a diameter of more than 7.5 mm show a better gastric residence time compared with one having 9.9 mm.
Table 3: Effect of tablet size on Gastric Emptying time

1.2.3.5 Density
The density of a dosage form also affects the gastric emptying rate. A buoyant dosage form having a density of less than that of the gastric fluids floats. Since it is away from the pyloric sphincter, the dosage unit is retained in the stomach for prolonged period. Timmermans et al (1994) studied the effect of buoyancy, posture and nature of meals on the gastric emptying process in vivo using gamma scintigraphy. To perform these studies, floating and non floating capsules of 3 different sizes having a diameter of 4.8 mm (small units), 7.5 mm (medium units), and 9.9 mm (large units), were formulated. On comparison of floating and nonfloating dosage units, it was concluded that regardless of their sizes the floating dosage units remained buoyant on the gastric contents throughout their residence in the gastrointestinal tract, while the nonfloating dosage units sank and remained in the lower part of the stomach. Floating units away from the gastro-duodenal junction were protected from the peristaltic waves during digestive phase while the nonfloating forms stayed close to the pylorus and were subjected to propelling and retropelling waves of the digestive phase. It was also observed that of the floating and nonfloating units, the floating units were had a longer gastric residence time for small and medium units while no significant difference was seen between the 2 types of large unit dosage forms.
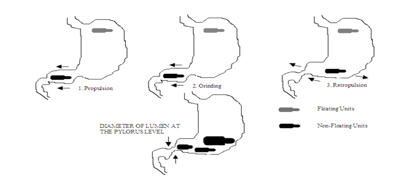
When subjects were kept in the supine position it was observed that the floating forms could only prolong their stay because of their size; otherwise the buoyancy remained no longer an advantage for gastric retention. A comparison was made to study the affect of fed and nonfed stages on gastric emptying. For this study all subjects remaining in an upright position were given a light breakfast and another similar group was fed with a succession of meals given at normal time intervals. It was concluded that as meals were given at the time when the previous digestive phase had not completed, the floating form buoyant in stomach could retain its position for another digestive phase as it was carried by the peristaltic waves in the upper part of the stomach.
1.3 DIFFERENT TECHNIQUES USED FOR GASTRIC RETENTION
The following approaches have been used for the design of gastro-retentive dosage forms
1.3.1 Single-Unit Dosage Forms
In Low-density approach the globular shells apparently having lower density than that of gastric fluid can be used as a carrier for drug for its controlled release. A buoyant dosage form can also be obtained by using a fluid-filled system that floating the stomach. In coated shells popcorn, poprice, and polystyrol have been exploited as drug carriers. Sugar polymeric materials such as methacrylic polymer and cellulose acetate phthalate have been used to undercoat these shells. They are further coated with a drug-polymer mixture. The polymer of choice can be either ethylcellulose or hydroxypropyl cellulose depending on the type of release desired. Finally, the product floats on the gastric fluid while releasing the drug gradually over a prolonged duration.Fluid- filled floating chamber type of dosage forms includes incorporation of a gas-filled floatation chamber into a microporous component that houses a drug reservoir. Apertures or openings are present along the top and bottom walls through which the gastrointestinal tract fluid enters to dissolve the drug. The other two walls in contact with the fluid are sealed so that the undissolved drug remains therein. The fluid present could be air, under partial vacuum or any other suitable gas, liquid, or solid having an appropriate specific gravity and an inert behavior. The device is of swallowable size, remains afloat within the stomach for a prolonged time, and the complete release the shell disintegrates, passes off to the intestine, and is eliminated.
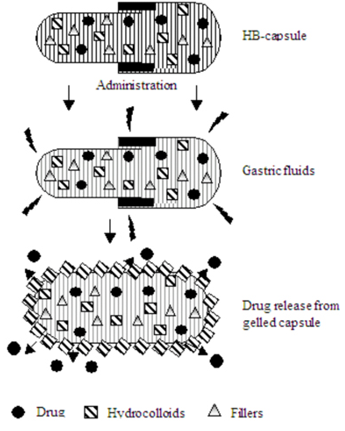
Hydrodynamically balanced systems (HBS) are designed to prolong the stay of the dosage form in the gastro intestinal tract and aid in enhancing the absorption. Such systems are best suited for drugs having a better solubility in acidic environment and also for the drugs having specific site of absorption in the upper part of the small intestine. To remain in the stomach for a prolongedperiod of time the dosage form must have a bulk density of less than 1, it should stay in the stomach, maintain its structural integrity, and drug constantly from the dosage form. The success of HBS capsule as a better system is best exemplified with chlordiazeopoxide hydrochloride. The drug is a classical example of a solubility problem wherein it exhibits a 4000-fold difference in solubility going from pH 3 to 6 (the solubility of chlordiazepoxide hydrochloride is 150 mg/mL and is ~0.1 mg/mL at neutral pH).mg commercial capsules. HBS can either be formulated as a floating tablet or capsule. Many polymers and polymer combinations with wet granulation as a manufacturing technique have been explored to yield floatable tablets.Various types of tablets (bilayered and matrix) have been shown to have floatable characteristics. Some of the polymers used are hydroxypropyl cellulose, hydroxypropyl methylcellulose, crosspovidone, sodium carboxymethyl cellulose, and ethyl cellulose. Self-correcting floatable asymmetric configuration drug delivery system23employs a disproportionate 3-layer matrix technology to control drugrelease.The 3-layer principle has been improved by development of an asymmetric configuration drug delivery system in order to modulate the release extent and achieve zero-order release kinetics by initially maintaining a constant area at the diffusing front with subsequent dissolution/erosion toward the completion of the release process. The system was designed in such a manner that it floated to prolong gastric residence time in vivo, resulting in longer total transit time within the gastrointestinal tract environment with maximum absorptive capacity and consequently greater bioavailability. This particular characteristic would be applicable to drugs that have pH-dependent solubility, a narrow window of absorption, and are absorbed by active transport from either the proximal or distal portion of the small intestine Single-unit formulations are associated with problems such as sticking together or being obstructed in the gastrointestinal tract, which may have a potential danger of producing irritation.
1.3.2 Multiple-Unit Dosage Forms
The purpose of designing multiple-unit dosage form is to develop a reliable formulation that has all the advantages of a single-unit form and also is devoid of any of the above mentioned disadvantages of single-unit formulations. In pursuit of this endeavor many multiple-unit floatable dosage forms have been designed. Microspheres have high loading capacity and many polymers have been used such as albumin, gelatin, starch, polymethacrylate, polyacrylamine, and polyalkylcyanoacrylate. Spherical polymeric microsponges, also referred to as “microballoons,” have been prepared. Microspheres have a characteristic internal hollow structure and show an excellent in vitro floatability. In Carbon dioxide–generating multiple-unit oral formulations several devices with features that extend, unfold, or are inflated by carbon dioxide generated in the devices after administration have been described in the recent patent literature. These dosage forms are excluded from the passage of the pyloric sphincter if a diameter of ~12 to 18 mm in their expanded state is exceeded.
1.3.3 Classification of Floating Drug Delivery Systems (FDDS):
Floating drug delivery systems are classified depending on the use of 2 formulation variables: effervescent and non-effervescent systems.
1.3.3.1 Effervescent Floating Dosage Forms
These are matrix types of systems prepared with the help of swellable polymers such as methylcellulose and chitosan and various effervescent compounds, eg, sodium bicarbonate, tartaric acid, and citric acid. They are formulated in such a way that when in contact with the acidic gastric contents, CO2 is liberated and gets entrapped in swollen hydrocolloids, which provides buoyancy to the dosage forms.
Ichikawa et al (1991) developed a new multiple type of floating dosage system composed of effervescent layers and swellable membrane layers coated on sustained release pills. The inner layer of effervescent agents containing sodium bicarbonate and tartaric acid was divided into 2 sublayers to avoid direct contact between the 2 agents. These sublayers were surrounded by a swellable polymer membrane containing polyvinyl acetate and purified shellac.
When this system was immersed in the buffer at 37ºC, it settled down and the solution permeated into the effervescent layer through the outer swellable membrane. CO2 was generated by the neutralization reaction between the 2 effervescent agents, producing swollen pills (like balloons) with a density less than 1.0 g/mL. It was found that the system had good floating ability independent of pH and viscosity and the drug (para-amino benzoic acid) released in a sustained manner.
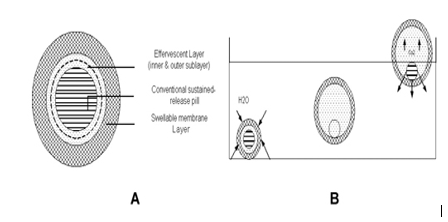
Ichikawa et al (1989) developed floating capsules composed of a plurality of granules that have different residence times in the stomach and consist of an inner foamable layer of gas-generating agents. This layer was further divided into 2 sublayers, the outer containing sodium bicarbonate and the inner containing tartaric acid. This layer was surrounded by an expansive polymeric film (composed of poly vinyl acetate [PVA] and shellac), which allowed gastric juice to pass through, and was found to swell by foam produced by the action between the gastric juices and the gas-generating agents. It was shown that the swellable membrane layer played an important role in maintaining the buoyancy of the pills for an extended period of time. Two parameters were evaluated: the time for the pills to be floating (TPF) and rate of pills floating at 5 hours (FP5h). It was observed that both the TPF and FP5h increased as the percentage of swellable membrane layer coated on pills having a effervescent layer increased. As the percentage of swellable layer was increased from 13% to 25% (wt/wt), the release rate was decreased and the lag time for dissolution also increased. The percentage of swellable layer was fixed at 13% wt/wt and the optimized system showed excellent floating ability in vitro (TPF ~10 minutes and FP5h ~80%) independent of pH and viscosity of the medium.
Yang et al (1999) developed a swellable asymmetric triple-layer tablet with floating ability to prolong the gastric residence time of triple drug regimen (tetracycline, metronidazole, and clarithromycin) in Helicobacter pylori–associated peptic ulcers using hydroxy propyl methyl cellulose (HPMC) and poly (ethylene oxide) (PEO) as the rate-controlling polymeric membrane excipients. The design of the delivery system was based on the swellable asymmetric triple-layer tablet approach. Hydroxypropylmethylcellulose and poly(ethylene oxide) were the major rate-controlling polymeric excipients. Tetracycline and metronidazole were incorporated into the core layer of the triple-layer matrix for controlled delivery, while bismuth salt was included in one of the outer layers for instant release. The floatation was accomplished by incorporatinga gas-generating layer consisting of sodium bicarbonate: calcium carbonate (1:2 ratios) along with the polymers. The in vitro results revealed that the sustained delivery of tetracycline and metronidazole over 6 to 8 hours could be achieved while the tablet remained afloat. The floating feature aided in prolonging the gastric residence time of this system to maintain high-localized concentration of tetracycline and metronidazole.
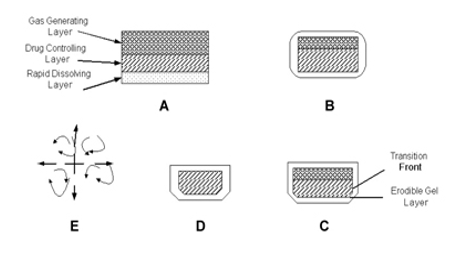
Ozdemir et al (2000) developed floating bilayer tablets with controlled release for furosemide. The low solubility of the drug could be enhanced by using the kneading method, preparing a solid dispersion with β cyclodextrin mixed in a 1:1 ratio. One layer contained the polymers HPMC 4000, HPMC 100, and CMC (for the control of the drug delivery) and the drug. The second layer contained the effervescent mixture of sodium bicarbonate and citric acid. The in vitro floating studies revealed that the lesser the compression force the shorter is the time of onset of floating, ie, when the tablets were compressed at 15 MPa, these could begin to float at 20 minutes whereas at a force of 32 MPa the time was prolonged to 45 minutes. Radiographic studies on 6 healthy male volunteers revealed that floating tablets were retained in stomach for 6 hours and further blood analysis studies showed that bioavailability of these tablets was 1.8 times that of the conventional tablets. On measuring the volume of urine the peak diuretic effect seen in the conventional tablets was decreased and prolonged in the case of floating dosage form.
Choi et al (2002) prepared floating alginate beads using gas-forming agents (calcium carbonate and sodium bicarbonate) and studied the effect of CO2 generation on the physical properties, morphology, and release rates. The study revealed that the kind and amount of gas-forming agent had a profound effect on the size, floating ability, pore structure, morphology, release rate, and mechanical strength of the floating beads. It was concluded that calcium carbonate formed smaller but stronger beads than sodium bicarbonate. Calcium carbonate was shown to be a less-effective gas-forming agent than sodium bicarbonate but it produced superior floating beads with enhanced control of drug release rates. In vitro floating studies revealed that the beads free of gas-forming agents sank uniformly in the media while the beads containing gas-forming agents in proportions ranging from 5:1 to 1:1 demonstrated excellent floating (100%).
Li et al (2001, 2002) evaluated the contribution of formulation variables on the floating properties of a gastro floating drug delivery system using a continuous floating monitoring device and statistical experimental design. The formulation was conceived using taguchi design. HPMC was used as a low-density polymer and citric acid was incorporated for gas generation. Analysis of variance (ANOVA) test on the results from these experimental designs demonstrated that the hydrophobic agent magnesium stearate could significantly improve the floating capacity of the delivery system. High-viscosity polymers had good effect on floating properties. The residual floating force values of the different grades of HPMC were in the order K4 M~ E4 M~K100 LV> E5 LV but different polymers with same viscosity, ie, HPMC K4M, HPMC E4M did not show any significant effect on floating property. Better floating was achieved at a higher HPMC/carbopol ratio and this result demonstrated that carbopol has a negative effect on the floating behavior.
Penners et al (1997) developed an expandable tablet containing mixture of polyvinyl lactams and polyacrylates that swell rapidly in an aqueous environment and thus reside in stomach over an extended period of time. In addition to this, gas-forming agents were incorporated. As the gas formed, the density of the system was reduced and thus the system tended to float on the gastric contents.
the tablet was found to swell on contact with aqueous medium. As the tablet dissolved, the barrier layers eroded away to expose more of the drug. Gas-evolving agent was added in either of the barrier layers, which caused the tablet to float and increased the retention of tablet in a patient’s stomach.
Talwar et al (2001) developed a once-daily formulation for oral administration of ciprofloxacin. The formulation was composed of 69.9% ciprofloxacin base, 0.34% sodium alginate, 1.03% xanthum gum, 13.7% sodium bicarbonate, and 12.1% cross-linked poly vinyl pyrrolidine. The viscolysing agent initially and the gel-forming polymer later formed a hydrated gel matrix that entrapped the gas, causing the tablet to float and be retained in the stomach or upper part of the small intestine (spatial control). The hydrated gel matrix created a tortuous diffusion path for the drug, resulting in sustained release of the drug (temporal delivery).
Two patents granted to Alza Corporation revealed a device having a hollow deformable unit that was convertible from a collapsed to expandable form and vice versa. The deformable unit was supported by a housing that was internally divided into 2 chambers separated by a pressure-sensitive movable bladder. The first chamber contained the therapeutic agent and the second contained a volatile liquid (cyclopentane, ether) that vaporized at body temperature and imparted buoyancy to the system. The system contained a bioerodible plug to aid in exit of the unit from the body (Michaels AS et al., 1975, 1974).
Baumgartner et al (2000) developed a matrix-floating tablet incorporating a high dose of freely soluble drug. The formulation containing 54.7% of drug, HPMC K4 M, Avicel PH 101, and a gas-generating agent gave the best results. It took 30 seconds to become buoyant. In vivo experiments with fasted state beagle dogs revealed prolonged gastric residence time. On radiographic images made after 30 minutes of administration, the tablet was observed in animal’s stomach and the next image taken at 1 hour showed that the tablet had altered its position and turned around. This was the evidence that the tablet did not adhere to the gastric mucosa. The MMC (phase during which large nondisintegrating particles or dosage forms are emptied from stomach to small intestine) of the gastric emptying cycle occurs approximately every 2 hours in humans and every 1 hour in dogs but the results showed that the mean gastric residence time of the tablets was 240 ± 60 minutes (n = 4) in dogs. The comparison of gastric motility and stomach emptying between humans and dogs showed no big difference and therefore it was speculated that the experimentally proven increased gastric residence time in beagle dogs could be compared with known literature for humans, where this time is less than 2 hours.
Moursy et al (2003) developed sustained release floating capsules of nicardipine HCl. For floating, hydrocolloids of high viscosity grades were used and to aid in buoyancy sodium bicarbonate was added to allow evolution of CO2. In vitro analysis of a commercially available 20-mg capsule of nicardipine HCl (MICARD) was performed for comparison. Results showed an increase in floating with increase in proportion of hydrocolloid. Inclusion of sodium bicarbonate increased buoyancy. The optimized sustained release floating capsule formulation was evaluated in vivo and compared with MICARD capsules using rabbits at a dose equivalent to a human dose of 40 mg. Drug duration after the administration of sustained release capsules significantly exceeded that of the MICARD capsules. In the latter case the drug was traced for 8 hours compared with 16 hours in former case.
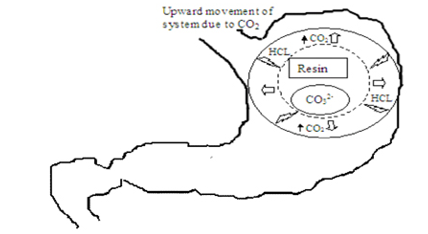
Atyabi and coworkers (1996) developed a floating system using ion exchange resin that was loaded with bicarbonate by mixing the beads with 1 M sodium bicarbonate solution. The loaded beads were then surrounded by a semipermeable membrane to avoid sudden loss of CO2. Upon coming in contact with gastric contents an exchange of chloride and bicarbonate ions took place that resulted in CO2 generation thereby carrying beads toward the top of gastric contents and producing a floating layer of resin beads.The in vivo behavior of the coated and uncoated beads was monitored using a single channel analyzing study in 12 healthy human volunteers by gamma radio scintigraphy. Studies showed that the gastric residence time was prolonged considerably (24 hours) compared with uncoated beads (1 to 3 hours).
1.3.3.2 Non-Effervescent Floating Dosage Forms
Non-effervescent floating dosage forms use a gel forming or swellable cellulose type of hydrocolloids, polysaccharides, and matrix-forming polymers like polycarbonate, polyacrylate, polymethacrylate, and polystyrene. The formulation method includes a simple approach of thoroughly mixing the drug and the gel-forming hydrocolloid. After oral administration this dosage form swells in contact with gastric fluids and attains a bulk density of < 1>
Thanoo et al (1993) developed polycarbonate microspheres by solvent evaporation technique. Polycarbonate in dichloromethane was found to give hollow microspheres that floated on water and simulated biofluids as evidenced by scanning electron microscopy (SEM). High drug loading was achieved and drug-loaded microspheres were able to float on gastric and intestinal fluids. It was found that increasing the drug-to-polymer ratio increased both their mean particle size and release rate of drug.
Nur and Zhang (2000) developed floating tablets of captopril using HPMC (4000 and 15 000 cps) and carbopol 934P. In vitro buoyancy studies revealed that tablets of 2 kg/cm2 hardness after immersion into the floating media floated immediately and tablets with hardness 4 kg/cm2 sank for 3 to 4 minutes and then came to the surface. Tablets in both cases remained floating for 24 hours. The tablet with 8 kg/cm2 hardness showed no floating capability. It was concluded that the buoyancy of the tablet is governed by both the swelling of the hydrocolloid particles on the tablet surface when it contacts the gastric fluids and the presence of internal voids in the center of the tablet (porosity). A prolonged release from these floating tablets was observed as compared with the conventional tablets and a 24-hour controlled release from the dosage form of captopril was achieved.
Bulgarelli et al (2000) studied the effect of matrix composition and process conditions on casein gelatin beads prepared by emulsification extraction method. Casein by virtue of its emulsifying properties causes incorporation of air bubbles and formation of large holes in the beads that act as air reservoirs in floating systems and serve as a simple and inexpensive material used in controlled oral drug delivery systems. It was observed that the percentage of casein in matrix increases the drug loading of both low and high porous matrices, although the loading efficiency of high porous matrices is lower than that of low porous matrices.
Fell et al (2000) prepared floating alginate beads incorporating amoxycillin. The beads were produced by dropwise addition of alginate into calcium chloride solution, followed by removal of gel beads and freeze-drying. The beads containing the dissolved drug remained buoyant for 20 hours and high drug-loading levels were achieved.
Streubel et al (2003) prepared single-unit floating tablets based on polypropylene foam powder and matrix-forming polymer. Incorporation of highly porous foam powder in matrix tablets provided density much lower than the density of the release medium. A 17% wt/wt foam powder (based on mass of tablet) was achieved in vitro for at least 8 hours. It was concluded that varying the ratios of matrix-forming polymers and the foam powder could alter the drug release patterns effectively.
Asmussen et al (2001) invented a device for the controlled release of active compounds in the gastrointestinal tract with delayed pyloric passage, which expanded in contact with gastric fluids and the active agent was released from a multiparticulate preparation. It was claimed that the release of the active compound was better controlled when compared with conventional dosage forms with delayed pyloric passage.
El-Kamel et al (2001) prepared floating microparticles of ketoprofen, by emulsion solvent diffusion technique. Four different ratios of Eudragit S 100 with Eudragit RL were used. The formulation containing 1:1 ratio of the 2 above-mentioned polymers exhibited high percentage of floating particles in all the examined media as evidenced by the percentage of particles floated at different time intervals. This can be attributed to the low bulk density, high packing velocity, and high packing factor.
Illum and Ping (2001) developed microspheres that released the active agent in the stomach environment over a prolonged period of time. The active agent was encased in the inner core of microspheres along with the rate-controlling membrane of a water-insoluble polymer. The outer layer was composed of bioadhesive (chitosan). The microspheres were prepared by spray drying an oil/water or water/oil emulsion of the active agent, the water-insoluble polymer, and the cationic polymer.
Streubel et al (2002) developed floating microparticles composed of
polypropylene foam, Eudragit S, ethyl cellulose (EC), and polymethyl metha acrylate (PMMA) and were prepared by solvent evaporation technique. High encapsulation efficiencies were observed and were independent of the theoretical drug loading. Good floating behavior was observed as more than 83% of microparticles were floating for at least 8 hours. The in vitro drug release was dependent upon the type of polymer used. At similar drug loading the release rates increased in the following order PMMA < EC>
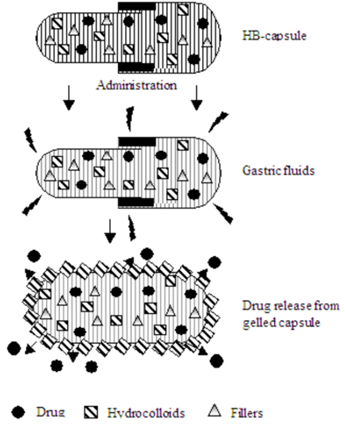
Sheth and Tossounian (1978) developed an HBS system containing a homogeneous mixture of drug and the hydrocolloid in a capsule, which upon contact with gastric fluid acquired and maintained a bulk density of less than 1 thereby being buoyant on the gastric contents of stomach until all the drug was released.
Ozdemir et al (2000) developed floating bilayer tablets with controlled release for furosemide. The low solubility of the drug could be enhanced by using the kneading method, preparing a solid dispersion with β cyclodextrin mixed in a 1:1 ratio. One layer contained the polymers HPMC 4000, HPMC 100, and CMC (for the control of the drug delivery) and the drug. The second layer contained the effervescent mixture of sodium bicarbonate and citric acid. The in vitro floating studies revealed that the lesser the compression force the shorter is the time of onset of floating, ie, when the tablets were compressed at 15 MPa, these could begin to float at 20 minutes whereas at a force of 32 MPa the time was prolonged to 45 minutes. Radiographic studies on 6 healthy male volunteers revealed that floating tablets were retained in stomach for 6 hours and further blood analysis studies showed that bioavailability of these tablets was 1.8 times that of the conventional tablets. On measuring the volume of urine the peak diuretic effect seen in the conventional tablets was decreased and prolonged in the case of floating dosage form.
Choi et al (2002) prepared floating alginate beads using gas-forming agents (calcium carbonate and sodium bicarbonate) and studied the effect of CO2 generation on the physical properties, morphology, and release rates. The study revealed that the kind and amount of gas-forming agent had a profound effect on the size, floating ability, pore structure, morphology, release rate, and mechanical strength of the floating beads. It was concluded that calcium carbonate formed smaller but stronger beads than sodium bicarbonate. Calcium carbonate was shown to be a less-effective gas-forming agent than sodium bicarbonate but it produced superior floating beads with enhanced control of drug release rates. In vitro floating studies revealed that the beads free of gas-forming agents sank uniformly in the media while the beads containing gas-forming agents in proportions ranging from 5:1 to 1:1 demonstrated excellent floating (100%).
Li et al (2001, 2002) evaluated the contribution of formulation variables on the floating properties of a gastro floating drug delivery system using a continuous floating monitoring device and statistical experimental design. The formulation was conceived using taguchi design. HPMC was used as a low-density polymer and citric acid was incorporated for gas generation. Analysis of variance (ANOVA) test on the results from these experimental designs demonstrated that the hydrophobic agent magnesium stearate could significantly improve the floating capacity of the delivery system. High-viscosity polymers had good effect on floating properties. The residual floating force values of the different grades of HPMC were in the order K4 M~ E4 M~K100 LV> E5 LV but different polymers with same viscosity, ie, HPMC K4M, HPMC E4M did not show any significant effect on floating property. Better floating was achieved at a higher HPMC/carbopol ratio and this result demonstrated that carbopol has a negative effect on the floating behavior.
Penners et al (1997) developed an expandable tablet containing mixture of polyvinyl lactams and polyacrylates that swell rapidly in an aqueous environment and thus reside in stomach over an extended period of time. In addition to this, gas-forming agents were incorporated. As the gas formed, the density of the system was reduced and thus the system tended to float on the gastric contents.
the tablet was found to swell on contact with aqueous medium. As the tablet dissolved, the barrier layers eroded away to expose more of the drug. Gas-evolving agent was added in either of the barrier layers, which caused the tablet to float and increased the retention of tablet in a patient’s stomach.
Talwar et al (2001) developed a once-daily formulation for oral administration of ciprofloxacin. The formulation was composed of 69.9% ciprofloxacin base, 0.34% sodium alginate, 1.03% xanthum gum, 13.7% sodium bicarbonate, and 12.1% cross-linked poly vinyl pyrrolidine. The viscolysing agent initially and the gel-forming polymer later formed a hydrated gel matrix that entrapped the gas, causing the tablet to float and be retained in the stomach or upper part of the small intestine (spatial control). The hydrated gel matrix created a tortuous diffusion path for the drug, resulting in sustained release of the drug (temporal delivery).
Two patents granted to Alza Corporation revealed a device having a hollow deformable unit that was convertible from a collapsed to expandable form and vice versa. The deformable unit was supported by a housing that was internally divided into 2 chambers separated by a pressure-sensitive movable bladder. The first chamber contained the therapeutic agent and the second contained a volatile liquid (cyclopentane, ether) that vaporized at body temperature and imparted buoyancy to the system. The system contained a bioerodible plug to aid in exit of the unit from the body (Michaels AS et al., 1975, 1974).
Baumgartner et al (2000) developed a matrix-floating tablet incorporating a high dose of freely soluble drug. The formulation containing 54.7% of drug, HPMC K4 M, Avicel PH 101, and a gas-generating agent gave the best results. It took 30 seconds to become buoyant. In vivo experiments with fasted state beagle dogs revealed prolonged gastric residence time. On radiographic images made after 30 minutes of administration, the tablet was observed in animal’s stomach and the next image taken at 1 hour showed that the tablet had altered its position and turned around. This was the evidence that the tablet did not adhere to the gastric mucosa. The MMC (phase during which large nondisintegrating particles or dosage forms are emptied from stomach to small intestine) of the gastric emptying cycle occurs approximately every 2 hours in humans and every 1 hour in dogs but the results showed that the mean gastric residence time of the tablets was 240 ± 60 minutes (n = 4) in dogs. The comparison of gastric motility and stomach emptying between humans and dogs showed no big difference and therefore it was speculated that the experimentally proven increased gastric residence time in beagle dogs could be compared with known literature for humans, where this time is less than 2 hours.
Moursy et al (2003) developed sustained release floating capsules of nicardipine HCl. For floating, hydrocolloids of high viscosity grades were used and to aid in buoyancy sodium bicarbonate was added to allow evolution of CO2. In vitro analysis of a commercially available 20-mg capsule of nicardipine HCl (MICARD) was performed for comparison. Results showed an increase in floating with increase in proportion of hydrocolloid. Inclusion of sodium bicarbonate increased buoyancy. The optimized sustained release floating capsule formulation was evaluated in vivo and compared with MICARD capsules using rabbits at a dose equivalent to a human dose of 40 mg. Drug duration after the administration of sustained release capsules significantly exceeded that of the MICARD capsules. In the latter case the drug was traced for 8 hours compared with 16 hours in former case.
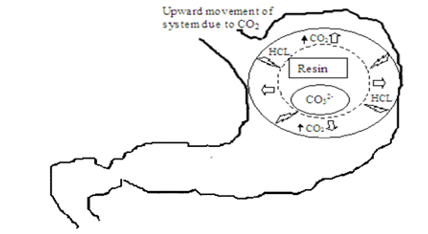
Atyabi and coworkers (1996) developed a floating system using ion exchange resin that was loaded with bicarbonate by mixing the beads with 1 M sodium bicarbonate solution. The loaded beads were then surrounded by a semipermeable membrane to avoid sudden loss of CO2. Upon coming in contact with gastric contents an exchange of chloride and bicarbonate ions took place that resulted in CO2 generation thereby carrying beads toward the top of gastric contents and producing a floating layer of resin beads.The in vivo behavior of the coated and uncoated beads was monitored using a single channel analyzing study in 12 healthy human volunteers by gamma radio scintigraphy. Studies showed that the gastric residence time was prolonged considerably (24 hours) compared with uncoated beads (1 to 3 hours).
1.3.3.2 Non-Effervescent Floating Dosage Forms
Non-effervescent floating dosage forms use a gel forming or swellable cellulose type of hydrocolloids, polysaccharides, and matrix-forming polymers like polycarbonate, polyacrylate, polymethacrylate, and polystyrene. The formulation method includes a simple approach of thoroughly mixing the drug and the gel-forming hydrocolloid. After oral administration this dosage form swells in contact with gastric fluids and attains a bulk density of < 1>
Thanoo et al (1993) developed polycarbonate microspheres by solvent evaporation technique. Polycarbonate in dichloromethane was found to give hollow microspheres that floated on water and simulated biofluids as evidenced by scanning electron microscopy (SEM). High drug loading was achieved and drug-loaded microspheres were able to float on gastric and intestinal fluids. It was found that increasing the drug-to-polymer ratio increased both their mean particle size and release rate of drug.
Nur and Zhang (2000) developed floating tablets of captopril using HPMC (4000 and 15 000 cps) and carbopol 934P. In vitro buoyancy studies revealed that tablets of 2 kg/cm2 hardness after immersion into the floating media floated immediately and tablets with hardness 4 kg/cm2 sank for 3 to 4 minutes and then came to the surface. Tablets in both cases remained floating for 24 hours. The tablet with 8 kg/cm2 hardness showed no floating capability. It was concluded that the buoyancy of the tablet is governed by both the swelling of the hydrocolloid particles on the tablet surface when it contacts the gastric fluids and the presence of internal voids in the center of the tablet (porosity). A prolonged release from these floating tablets was observed as compared with the conventional tablets and a 24-hour controlled release from the dosage form of captopril was achieved.
Bulgarelli et al (2000) studied the effect of matrix composition and process conditions on casein gelatin beads prepared by emulsification extraction method. Casein by virtue of its emulsifying properties causes incorporation of air bubbles and formation of large holes in the beads that act as air reservoirs in floating systems and serve as a simple and inexpensive material used in controlled oral drug delivery systems. It was observed that the percentage of casein in matrix increases the drug loading of both low and high porous matrices, although the loading efficiency of high porous matrices is lower than that of low porous matrices.
Fell et al (2000) prepared floating alginate beads incorporating amoxycillin. The beads were produced by dropwise addition of alginate into calcium chloride solution, followed by removal of gel beads and freeze-drying. The beads containing the dissolved drug remained buoyant for 20 hours and high drug-loading levels were achieved.
Streubel et al (2003) prepared single-unit floating tablets based on polypropylene foam powder and matrix-forming polymer. Incorporation of highly porous foam powder in matrix tablets provided density much lower than the density of the release medium. A 17% wt/wt foam powder (based on mass of tablet) was achieved in vitro for at least 8 hours. It was concluded that varying the ratios of matrix-forming polymers and the foam powder could alter the drug release patterns effectively.
Asmussen et al (2001) invented a device for the controlled release of active compounds in the gastrointestinal tract with delayed pyloric passage, which expanded in contact with gastric fluids and the active agent was released from a multiparticulate preparation. It was claimed that the release of the active compound was better controlled when compared with conventional dosage forms with delayed pyloric passage.
El-Kamel et al (2001) prepared floating microparticles of ketoprofen, by emulsion solvent diffusion technique. Four different ratios of Eudragit S 100 with Eudragit RL were used. The formulation containing 1:1 ratio of the 2 above-mentioned polymers exhibited high percentage of floating particles in all the examined media as evidenced by the percentage of particles floated at different time intervals. This can be attributed to the low bulk density, high packing velocity, and high packing factor.
Illum and Ping (2001) developed microspheres that released the active agent in the stomach environment over a prolonged period of time. The active agent was encased in the inner core of microspheres along with the rate-controlling membrane of a water-insoluble polymer. The outer layer was composed of bioadhesive (chitosan). The microspheres were prepared by spray drying an oil/water or water/oil emulsion of the active agent, the water-insoluble polymer, and the cationic polymer.
Streubel et al (2002) developed floating microparticles composed of
polypropylene foam, Eudragit S, ethyl cellulose (EC), and polymethyl metha acrylate (PMMA) and were prepared by solvent evaporation technique. High encapsulation efficiencies were observed and were independent of the theoretical drug loading. Good floating behavior was observed as more than 83% of microparticles were floating for at least 8 hours. The in vitro drug release was dependent upon the type of polymer used. At similar drug loading the release rates increased in the following order PMMA < EC>
Sheth and Tossounian (1978) developed an HBS system containing a homogeneous mixture of drug and the hydrocolloid in a capsule, which upon contact with gastric fluid acquired and maintained a bulk density of less than 1 thereby being buoyant on the gastric contents of stomach until all the drug was released.
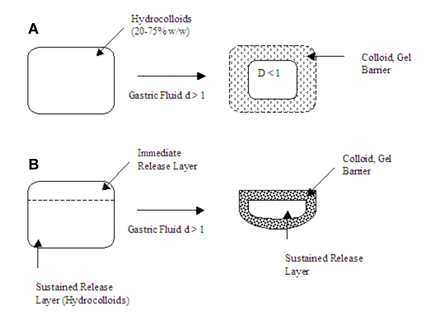
Sheth and Tossounian (1979) developed hydrodynamically balanced sustained release tablets containing drug and hydrophilic hydrocolloids, which on contact with gastric fluids at body temperature formed a soft gelatinous mass on the surface of the tablet and provided a water-impermeable colloid gel barrier on the surface of the tablets. The drug slowly released from the surface of the gelatinous mass that remained buoyant on gastric fluids (Figure 8, A and B).
Ushomaru et al (1987) developed sustained release composition for a capsule containing mixture of cellulose derivative or a starch derivative that formed a gel in water and higher fatty acid glyceride and/or higher alcohol, which was solid at room temperature. The capsules were filled with the above mixture and heated to a temperature above the melting point of the fat components and then cooled and solidified.
Bolton and Desai (1989) developed a noncompressed sustained release tablet that remained afloat on gastric fluids. The tablet formulation comprised 75% of drug and 2% to 6.5% of gelling agent and water. The noncompressed tablet had a density of less than 1 and sufficient mechanical stability for production and handling.
Kawashima et al (1991) prepared multiple-unit hollow microspheres by emulsion solvent diffusion technique. Drug and acrylic polymer were dissolved in an ethanol-dichloromethane mixture, and poured into an aqueous solution of PVA with stirring to form emulsion droplets. The rate of drug release in micro balloons was controlled by changing the polymer-to-drug ratio. Microballoons were floatable in vitro for 12 hours when immersed in aqueous media. Radiographical studies proved that microballoons orally administered to humans were dispersed in the upper part of stomach and retained there for 3 hours against peristaltic movements.
Dennis et al (1992) invented a buoyant controlled release pharmaceutical powder formulation filled into capsules. It released a drug of a basic character at a controlled rate regardless of the pH of the environment. PH-dependent polymer is a salt of a polyuronic acid such as alginic acid and a pH-independent hydrocarbon gelling agent, hydroxypropylmethyl cellulose.
Spickett et al (1993) invented an antacid preparation having a prolonged gastric residence time. It comprised 2 phases. The internal phase consisted of a solid antacid and the external phase consisted of hydrophobic organic compounds (mono-, di-, and triglycerides) for floating and a non-ionic emulsifier.
Franz and Oth (1993) described a sustained release dosage form adapted to release of the drug over an extended period of time. It comprised a bilayer formulation in which one layer consisted of drug misoprostal and the other had a floating layer. The uncompressed bilayer formulation was kept in a capsule and was shown to be buoyant in the stomach for 13 hours. The dosage form was designed in such a way that the entire drug was released in the stomach itself.
Wu et al (1997) developed floating sustained release tablets of nimodipine by using HPMC and PEG 6000. Prior to formulation of floating tablets, nimodipine was incorporated into poloxamer-188 solid dispersion after which it was directly compressed into floating tablets. It was observed that by increasing the HPMC and decreasing the PEG 6000 content a decline in vitro release of nimodipine occurred.
Wong et al (2000) developed a prolonged release dosage form adapted for gastric retention using swellable polymers. It consisted of a band of insoluble material that prevented the covered portion of the polymer matrix from swelling and provided a segment of a dosage form that was of sufficient rigidity to withstand the contractions of the stomach and delayed the expulsion of the dosage form from the stomach.
Harrigan (1977) developed an intragastric floating drug delivery system that was composed of a drug reservoir encapsulated in a microporous compartment having pores on top and bottom surfaces. However, the peripheral walls were sealed to prevent any physical contact of the drug in the reservoir with the stomach walls.
Joseph et al (2002) developed a floating dosage form of piroxicam based on hollow polycarbonate microspheres. The microspheres were prepared by the solvent evaporation technique. Encapsulation efficiency of ~95% was achieved. In vivo studies were performed in healthy male albino rabbits. Pharmacokinetic analysis was derived from plasma concentration vs time plot and revealed that the bioavailability from the piroxicam microspheres alone was 1.4 times that of the free drug and 4.8 times that of a dosage form consisting of microspheres plus the loading dose and was capable of sustained delivery of the drug over a prolonged period.
Table 4: Drugs Reported To Be Used In The Formulation Of Floating Dosage Forms:
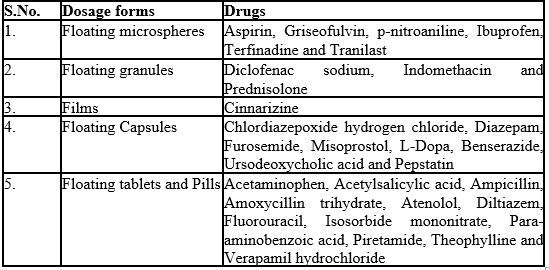
Table 5: Marketed preparation:
1.3.3.3 Bioadhesive systems:
This approach involves the use of bioadhesive polymers, which can adhere to the epithelial surface in the stomach. The original concept of bioadhesive polymers as platforms for oral controlled drug delivery was to use these polymers to control and to prolong the GI transit of oral controlled delivery systems for all kinds of drugs. Whereas bioadhesion has found interesting applications for other routes of administration (buccal, nasal, rectal and vaginal), it now seems that the controlling approach of GI transit has been abandoned before having shown any significant clinical outcome [17].
According to in vivo results obtained in animals and in humans, it does not seem that mucoadhesive polymers are able to control and slow down significantly the GI transit of solid delivery systems. Attention should be paid to possible occurrence of local ulcerous side effects due to the intimate contact of the system with mucosa for prolonged periods of time. The continuous production of mucous by the gastric mucosa to replace the mucous that is lost through peristaltic contractions and the dilution of the stomach content also seems to limit the potential of mucoadhesion as a gastroretentive force [18].
1.3.3.4 High Density Systems:
Sedimentation has been employed as a retention mechanism for pellets that are small enough to be retained in the folds of stomach body near the pyloric region, which is the part of the organ with the lowest position in an upright posture. Dense pellets (approximately 3g/cm3) trapped in fold also tend to withstand the peristaltic movements of the stomach wall.With pellets,the GI transit time can be extended from an average of 5.8 -25 hours, depending more on density than on diameter of the pallets. Commonly used excipients are barium sulphate, Zinc, Oxide,titanium dioxide and iron powder, etc. These materials increase density by up 1.5-2.4g/cm-3.
RATIONALE FOR DEVELOPING CIPROFLOXACIN FLOATING TABLETS
Ciprofloxacin is a member of Fluroquinoline group of antibiotic, used in clinical management of patients with Bacterial infection, The elimination half life of Ciprofloxacin is 7 hrs which indicated its suitability in formulating into a sustained release dosage form. The oral bioavailability of Ciprofloxacin has been reported to be 60%.
Due to its high solubility in acidic retention of Ciprofloxacin may offer numerous advantages, including, increase in the extent of absorption, improved bio-availability and therapeutic efficacy. Frequent administration of Ciprofloxacin (100mg b.i.d/t.i.d) also prompted to make floating sustained release tablets of Ciprofloxacin. Based on this, an attempt was made through this investigation to formulate floating matrix tablets of Ciprofloxacin using different polymers.
The solubility and stability of Ciprofloxacin in hydrochloric acid helps for better absorption in acidic environment. By employing gastro-retentive floating drug delivery systems, the dosage form is retained in the stomach and the drug is released in a controlled fashion.
1.5 BJECTIVE
The present research work aims to formulate and evaluate sustained release floating matrix tablets of Ciprofloxacin using combination of different polymers.
2. PLAN OF WORK
To achieve the mentioned objective, the experimental study was framed as below:
- Dose calculation for sustained release of Ciprofloxacin for 12 hours.
- Formulation of floating tablets of Ciprofloxacin using HPMC K4M &HPMC K15M
- Evaluation of the final blend for Compressibility index, Hausner ratio and Angle of repose
- Evaluation of the prepared floating tablets:
i) The matrix tablets are characterized for the following parameters:
Weight variation
Thickness
Hardness
Friability
Drug content uniformity
Floating lag time
Floating duration time
ii) In vitro dissolution study of Floating tablets.
3. MATERIALS AND METHODS
TABLE 6: MATERIALS USED

TABLE 7: EQUIPMENT USED
4. DRUG PROFILE
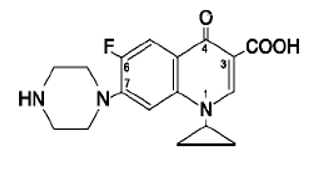
Ciprofloxacin:
- Ciprofloxacin is a first generation fluoroquinolone broad spectrum antibiotic
- IUPAC name: 1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)- 3-quinolinecarboxylic acid.
- Empirical formula : C17H18FN3O3
- Molecular weight : 331.4 g/mole
- Description: It is a faintly yellowish to light yellow crystalline substance.
- chemical structure is as follows:
- Bioavailability : 69%[1]
- Metabolism : Hepatic, including CYP1A2
- Half-life : 4 hours
- Excretio : Renal
Mode of action:
Ciprofloxacin is a broad-spectrum antibiotic active against both Gram-positive and Gram-negative bacteria. It functions by inhibiting DNA gyrase, a type II topoisomerase, and topoisomerase IV,[40] enzymes necessary to separate bacterial DNA, thereby inhibiting cell division.
Uses:
C iprofloxacin is used in the treatment of :
- Urinary Tract Infections caused by Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Serratia marcescens, Proteus mirabilis, Providencia rettgeri, Morganella morganii, Citrobacter diversus, Citrobacter freundii, Pseudomonas aeruginosa, Staphylococcus epidermidis, Staphylococcus saprophyticus, or Enterococcus faecalis.
- Acute Uncomplicated Cystitis in females caused by Escherichia coli or Staphylococcus saprophyticus.
- Chronic Bacterial Prostatitis caused by Escherichia coli or Proteus mirabilis.
- Lower Respiratory Tract Infections caused by Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Proteus mirabilis, Pseudomonas aeruginosa, Haemophilus influenzae, Haemophilus parainfluenzae, or Streptococcus pneumoniae. Also, Moraxella catarrhalis for the treatment of acute exacerbations of chronic bronchitis.
- Acute Sinusitis caused by Haemophilus influenzae, Streptococcus pneumonia and catarrhalis.
- Skin and Skin Structure Infections caused by Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Proteus mirabilis, Proteus vulgaris, Providencia stuartii, Morganella morganii, Citrobacter freundii, Pseudomonas aeruginosa, Staphylococcus aureus (methicillin-susceptible), Staphylococcus epidermidis, or Streptococcus pyogenes.
- Bone and Joint Infections caused by Enterobacter cloacae, Serratia marcescens, or Pseudomonas aeruginosa.
- Complicated Intra-Abdominal Infections (used in combination with metronidazole) caused by Escherichia coli, Pseudomonas aeruginosa, Proteus mirabilis, Klebsiella pneumoniae, or Bacteroides fragilis.
- Infectious Diarrhea caused by Escherichia coli (enterotoxigenic strains), Campylobacter jejuni, Shigella boydii **/* , Shigella dysenteriae, Shigella flexneri or Shigella sonnei **/* when antibacterial therapy is indicated.
- Typhoid Fever (Enteric Fever) caused by Salmonella typhi.
- Uncomplicated cervical and urethral gonorrhea due to Neisseria gonorrhoeae.
- Complicated Urinary Tract Infections and Pyelonephritis due to Escherichia coli.
- EXCIPIENT PROFILE
An excipient is generally a pharmacologically inactive substance used as a carrier for the active ingredients of a medication
5.1 HYDROXY PROPYL METHYL CELLULOSE (HPMC)
Hydroxy propyl methylcellulose is mixed alkyl hydroxyl alkyl cellulosic ether and may be regarded as the propylene glycol ether of methylcellulose.
Chemical name: Cellulose, 2-hydroxy propyl methyl ether
Common name: Methocel, Hypromellose
Grades: HPMC M
Description: It is an odorless, tasteless, white or creamy white fibrous granular powder. Solubility: Soluble in cold water, forming a viscous colloidal solution, insoluble in alcohol, ether and chloroform.
Gel point: 500C to 900C
pH: 6.0 to 8.0 (1% aqueous solution)
scosity: 80-120(K100LV), 11250-21000(K15M), 80000-120000 cps (K100M)
Very stable in dry conditions, solutions are stable at pH3.0-11.0. Aqueous solutions are liable to be effected by microbes.
Uses: Suspending agent, viscosity modifier, film and matrix forming material. Tablet binder and adhesive ointment ingredient.
5.2. SODIUM BICARBONATE
Synonyms:Sodium hydrogen carbonate, Baking soda
ption: Occurs as an odorless, white crystalline powder with a saline taste. The crystalline structure is monoclinic prisms.
Solubility: Soluble in water, practically insoluble in ethanol and ether.
Functional Category: Alkalizing agent, therapeutic agent
Application in pharmaceutical technology:
Sodium bicarbonate is generally used in pharmaceutical formulations as a source of carbondioxide in effervescent tablets and granules. In effervescent tablets and granules, it is formulated with either citric acid or tartaric acid. It is used as buffering agent. Therapeutically it is used as antacid and as a source of the bicarbonate anion in the treatment of metabolic acidosis. It is also used as a component of oral rehydration salts. It is used in food products as leavening agent.
Use Concentration (%)
Buffered tablets 10-40
Effervescent tablets 25-40
Isotonic injection/infusion 1.39
5.3. LACTOSE
Lactose is a carbohydrate, and as such a disaccharide. One Molecule of lactose consists of one molecule each of two other carbohydrates, i.e. galactose and glucose. These galactose and glucose moieties, as they are called, are linked together by means of what is known as a beta-(1,4) glucosidic linkage. The molecular structure of lactose is depicted below.
- Chemical name : 4-O-β-D-galactopyranosyl, D-glucopyranose.
- Physicochemical properties
- Taste, palatability, sweetness : Lactose has a clean and sweet taste without any aftertaste. The sweetness profile resembles that of sucrose. However, the relative sweetness of lactose is small (only 20%) when compared to sucrose (100%). β-lactose appears to be somewhat sweeter than α-lactose, probably due to the fact that β-lactose dissolves somewhat quicker in the saliva of the mouth than α-lactose, hence reaching a higher concentration in the same period of time and thus giving rise to a higher sweetness sensation.
Typical values of Bulk Density, Tapped Density, Hausner ratio and Carr’s Index
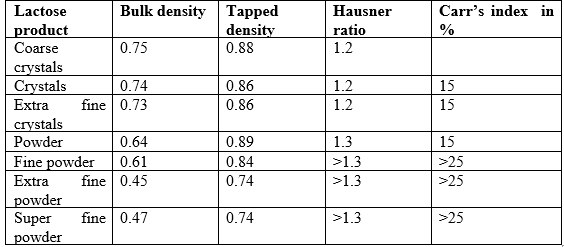
- Solubility :
Lactose is freely soluble in water. Solubility increases with increasing temperature.
β-lactose dissolves more readily than α-lactose, as is apparent from their very different initial rates of solubility.The particle size of the lactose influences its dissolving velocity. Coarse lactose crystals dissolve much slower than tiny lactose particles. Dissolving velocity, and hence particle size, does not alter final solubility.
- Uses:
Used as
- filler in direct compression of tablets
- micronized drug carrier
- binder
5.4. MAGNESIUM STEARATE:
Synonyms: magnesium salt
Description:It is a slight odored white substance which is solid at room temperature. It is a salt containing two equivalents of stearate (the anion of stearic acid) and one magnesium cation (Mg2+)
ng point : 88 °C, 361 K, 190 °F
Solubility: Solubility in water is negligible.Insoluble in ether and slightly soluble in benzene.
Chemical structure:
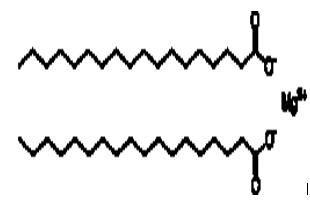
IUPAC name: magnesium octadecanoate
Chemical formula : Mg(C18H35O2)2
Molecular mass : 591.27 g/mol
Uses: used as lubricant and diluents
STANDARD GRAPH OF CIPROFLOXACIN
An accurately weighed amount of 100mg Ciprofloxacin was transferred into a 100 ml volumetric flask containing 0.1N HCl to dissolve and then the volume was made up to the mark with 0.1N HCl. From this necessary dilutions were made to give concentration ranging from 1-32 µg/ml solutions. The absorbance of the volumetric solutions was recorded at λmax (272nm) of the drug and plotted graphically to give the standard graph of Ciprofloxacin
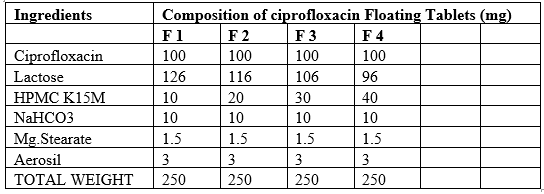
5.6 PREPARATION OF CIPROFLOXACIN FLOATING TABLETS
The Compositions of different formulation trials with different polymers. Accurately weighed quantities of polymer, avicel were taken in a mortar and mixed geometrically. To this mixture required quantity of Ciprofloxacin was added and mixed slightly with pestle. This mixture was passed through 40# and later collected in a plastic bag and blended for 5 min. To this required amount of sodium bi carbonate was added and again mixed for 5 min. Later required quantity of magnesium stearate and aerosol were added and the final blend was again passed through 40#. Thus obtained blend was mixed thoroughly for 10 min and compressed into tablets with 6mm Round Punches and corresponding dies at a hardness of 6kg/cm2 on a rotary tablet punching machine
6. EVALUATION OF FINAL POWDER BLEND
The powder blend of all formulations was evaluated for Bulk density, Tapped density, Compressibility Index, Hausner ratio and Angle of repose.
- Bulk Density
30gms of material was passed through a sieve no. 25 to break up agglomerates and introduced into a dry 100mL cylinder, without compacting, the powder was carefully leveled without compacting and the unsettled apparent volume, Vo, was read. The bulk density was calculated, in grams per ml, using the formula (Shah et al., 1997).
(M) / (Vo)
Where M = Total weight of the powder blend and V0 is the bulk volume of the powder blend
B) Tapped Density
After carrying out the procedure as given in the measurement of bulk density the cylinder containing the sample was tapped using a mechanical tapped density tester (Electrolab) that provides a fixed drop of 14±2 mm at a nominal rate of 300 drops per minute. The cylinder was tapped 500 times initially followed by an additional tap of 750 times until difference between succeeding measurement was less than 2% and then tapped volume Vf, was measured to the nearest graduated unit. The tapped density was calculated, in g per ml, using the formula:
(M) / (Vf)
Where M = Total weight of the powder blend and Vf is the tapped volume of the powder blend
C) Measures of Powder Compressibility
The Compressibility Index and Hausner Ratio are measures of the propensity of a powder to be compressed. As such, they are measures of the relative importance of inter particulate interactions. As such, they are measures of the relative importance of inter particulate interactions. In a free-flowing powder, such interactions are generally less significant, and the bulk and tapped densities will be closer in value. For poorer flowing materials, there are frequently greater interparticle interactions and a greater difference between the bulk and tapped densities will be observed. These differences are reflected in the Compressibility Index and the Hausner Ratio, which are calculated using the following formulae:
Compressibility Index = (Vr-Vo) * 100 / Vr
Where , Vr = Tapped density ; Vo = Bulk density
D) Hausner Ratio :
It is the ratio of bulk density to tapped density
Vo/ Vf
Vo = Bulk density; Vr= Tapped density
E) Angle of Repose
The fixed funnel method was employed to measure the repose angle. A funnel was secured with its tip at a given height, H above a graph paper that was placed on a flat horizontal surface. The blend was carefully pored through the funnel until the apex of the conical pile just touched the tip of the funnel. The radius, R, of the base of the conical pile was measured. The angle of repose, α, was calculated using the following formula: (Copper J, et al., 1986).
α = tan-1 H/R
6.1 DETERMINATION OF PHYSICAL PARAMETERS OF FLOATING TABLETS
6.1.1Weight Variation test
Twenty (20) tablets from each batch were individually weighed in grams on an analytical balance. The average weight and standard deviation were calculated, individual weight of each tablet was also calculated using the same and compared with average weight
Weight Variation limits as per USP
Average weight in mg. % ± deviation allowed
130 or less 10
- 7.5
More than 324 5.0
6.1.2 Thickness test
The thickness in millimeters (mm) was measured individually for 10 pre weighed tablets by using a Vernier Calipers. The average thickness and standard deviation were reported.
6.1.3 Hardness test
Tablet hardness was measured using a Monsanto hardness tester. The crushing strength of the 10 tablets with known weight and thickness of each was recorded in kg/cm2 and the average hardness, and the standard deviation was reported.
6.1.4 Friability test
Twenty (20) tablets were selected from each batch and weighed. Each group of tablets was rotated at 25 rpm for 4 minutes (100 rotations) in the Roche friabulator. The tablets were then dusted and re-weighed to determine the loss in weight. Friability was then calculated as per weight loss from the original tablets.
6.1.5 Determination of Drug Content
Ten tablets with pre determined weight from each batch were taken and crushed in a mortar and weight equivalent to one average tablet was taken, transferred to a 250 ml volumetric flask and 0.1N HCl was added. The volume was then made up to the mark with 0.1N HCl. The solution was filtered and the filtrate was sufficiently diluted and the absorbance was recorded against the blank at 272 nm. The drug content of the Standard containing the drug powder was also determined. The Drug content was determined by the formula.
Amount in test
Drug content = ------------------------------------ x 100
Amount in standard
The tablet passes the requirements if the amount of the active ingredient in each of the 10 tested tablets lies within the range of 85% to 115% of the stated amount.
3.1.6 In-vitro buoyancy Studies.
The in-vitro buoyancy (n= 3) was determined by floating lag times according to the method described by Rosa et al.The tablets were placed in a beaker containing 100 ml of 0.1N HCL. The time required for the tablet to rise to the surface and float was taken as floating lag time. Total floating time was also measured.
6.1.7 In vitro Drug Release Studies
The release rate of Ciprofloxacin floating tablets was determined using USP Type 2 Apparatus. The dissolution test was performed in triplicate, using 900ml of 0.1N HCL,at 37± 0.5˚C at 50 rpm for 4 hrs. A 5ml sample was withdrawn from the dissolution apparatus at specified time points and the samples were replaced with fresh dissolution medium.The samples were filtered through a 0.45-µm membrane filter and diluted if necessary. Absorbance of these solutions were measured at 272nm using Elico SL -159, U.V-Visible Spectrophotometer. Cumulative drug release was calculated using the equation (y = 0.03x + 0.024) generated from Beer Lambert’s Calibration curve in the linearity range of 1-32µ
7. DISSOLUTION PROFILES COMPARISION
In the development of oral controlled release preparations, an ethical or proprietary product, which has been available in the market and established its efficacy clinically, is usually selected as a reference. The generic preparation is always formulated with its dissolution profile as closely similar as possible to that of the proprietary product. Several methods have been proposed to compare the dissolution profiles of test with Reference (Fassihi and Pillay, 1998). Those methods were classified into several categories, such as:
a)Model Dependent methods & b) Model Independent methods
STANDARD GRAPH OF CIPROFLOXACIN
The standard graph of Ciprofloxacin in 0.1N HCl showed a good linearity with R2 of 0.999, in the concentration range of 0-32 μg/ml at 272nm
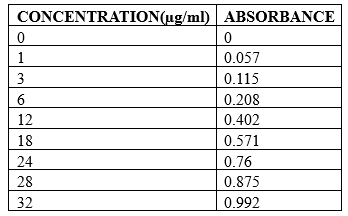
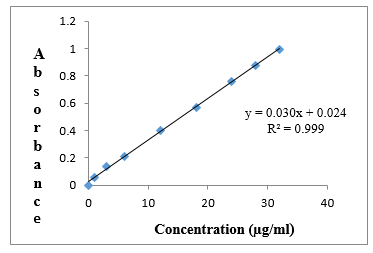
TABLE 16: Flow Properties of the Final Powder Blend
Formulation code | Compressibility index | Angle of repose | Hausner ratio |
F 1 | 12.3 | 28.7º | 1.15 |
F 2 | 15.9 | 29.3º | 1.19 |
F 3 | 12.8 | 27.6º | 1.13 |
F 4 | 15.7 | 28.1º | 1.17 |
All Formulations were evaluated for Compressibility index, Angle of repose and Hausner ratio. The results indicated the pre-compressed blend gas good flow.
7.1 EVALUATION OF THE PREPARED TABLETS FOR PHYSICAL
PARAMETERS.
All Formulations were tested for physical parameter like hardness, thickness, weight variation, friability and drug content. All estimated parameters were found to be within the limits.
TABLE 17: Physical Parameters of the Prepared Formulations
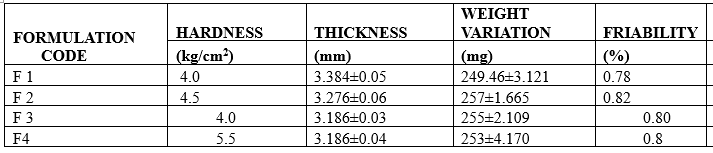
Table 18: Floating Properties of All Formulations
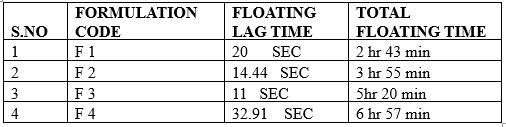
Tablets of all formulations had floating lag time below 23 seconds regardless of viscosity and content of HPMC because of evolution of CO2 resulting from the interaction between sodium bicarbonate and dissolution medium; entrapment of gas inside the hydrated polymeric matrices enables the dosage form to float by lowering the density of the matrices. It was reasoned that as for HPMC content of 10% or more, the particles of HPMC are close enough to permit a faster establishment of the gel layer inside which the CO2 gas gets entrapped leading to decreased density ultimately leading to floating of the tablet[15]. Floating time of all formulations was found to be less than 7 hrs.

IN-VITRO DRUG RELEASE OF FLOATING TABLETS
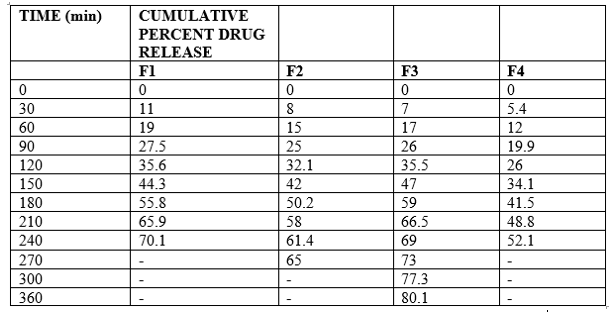
Results and Disscusions
The dissolution profiles of the formulations are represented graphically in and the results are shown in (table 19).The dissolution profile shows the cumulative percent drug release from the formulations having varying concentrations of polymer HPMC K15M . Formulation F 3 released 80.1% of the drug within 6 hrs. This was ascertained due to the insufficiency of the polymers to form a rigid gel barrier around the tablet ultimately leading to loss of matrix integrity. Increasing the polymer level (FH 4 formulation) resulted in release of only 52.1% of the total drug concentration pertaining to sustained release of drug .
8. Conclusion
Sustained release floating tablets of Ciprofloxacin were successfully prepared with hydrophilic polymers like HPMC K15M.
From the Preformulation studies for drug excipients compatibility, it was observed that no physical incompatibility existed between the drug and excipients.
The formulated batches were evaluated for physical parameters, floating properties and dissolution profiles. The physical properties like weight variation and friability of all formulations complied with the pharmacopoeial specifications. The drug content of all tablets was in the range of 98 – 102%.
The dissolution profile of formulation F3 is sustained release with 80.1% of drug release with in 6hrs.The floating time of F4 formulation is 6hrs 57min which shows enhanced bioavailability of ciprofloxacin.
References
- Hwang S. J., H. Park and K. Park, “Gastric Retentive Drug-Delivery Systems”, Crit. Rev. Ther. Drug Carrier Syst. 1998, 15 (3), 243–284.
View at Publisher | View at Google Scholar - Whitehead, J. T. Fell and J H Collett, “Development of a Gastroretentive Dosage Form”, Eur. J. Pharma. Sci., 1996, 4 (1),182.
View at Publisher | View at Google Scholar - Mojaverian, P. H. Vlasses, P. E. Kellner and M. . Rocci, “Effects of gender, posture and age on gastric residence time of an indigestible solid: pharmaceutical considerations”, Pharm. Res., 1988, 10, 639–644.
View at Publisher | View at Google Scholar - Deshpande, C.T. Rhodes, N.H. Shah,” controlled rlease drug delivery system for prolonged gastric residence: an overview,” Drug Dev.Ind. Pharm., 1996, 22, 531- 539.
View at Publisher | View at Google Scholar - Moses, “ Gastro retentive dosage forms,” Crit. Rev. Ther. Drug Carrier Syst.1993, 10, 143-195.
View at Publisher | View at Google Scholar - Whitelind, J.T. Fell, J.H. Collete, H.J. Sharma, “ An in vivo study demonstrating prolonged gastric retention”, J. Control. Rel..,1998, 55, 3-12.
View at Publisher | View at Google Scholar - Dave B.S, A.F.Amin and M.M. Patel “Gastroretentive drug delivery system of ranitidine hydrochloride formulation and in vitro evaluation” 2004,AAPS Pharm. Sci. Tech.,2004, 5(2),1-6.
View at Publisher | View at Google Scholar - Hwang S.K, H. Park, , K.Park,“Gastric retentive drug delivery systems.” Crit. Rev. Ther.Drug Carrier Syst., 1998, 15, 243–284.
View at Publisher | View at Google Scholar - Grubel et al, Gastric emptying of non-digestible solids in the fasted dog., J.Pharm.Sci.,1987, 76, 117 –122.
View at Publisher | View at Google Scholar - Gastro retentive drugs: a review, by Prahlad Tayade, Pharma Pulse Express
View at Publisher | View at Google Scholar - Desai and S. Bolton, “A Floating Controlled Release Drug Delivery System: In vitro–In vivo Evaluation”, Pharma. Res., 1993, 10 (9) 1321-325.
View at Publisher | View at Google Scholar - Singh B.M and K. H. Kim, “Floating drug delivery systems: an approach to controlled drug delivery via gastric retention”, J. Control. Rel., 2000, 63, 235–259.
View at Publisher | View at Google Scholar - Timmermans and A. J. Moes, “Factors controlling the buoyancy and gastric retention capabilities of floating matrix capsules: new data for reconsidering the controversy”, J. Pharm. Sci., 1994, 83, 8–24.
View at Publisher | View at Google Scholar - Timmermans and A. J. Moes, “How well do floating dosage forms float?”, Int. J.Pharm.,1990, 62, 207–216.
View at Publisher | View at Google Scholar - Seth and J. Tossounian, The hydrodynamically balanced system HBSTM: A novel drug deliverysystem for oral use, Drug Dev.Ind. Pharm. 1984,10, 313–339.
View at Publisher | View at Google Scholar - P. G Yeole., S. Khan, V. F Patel., Floating drug delivery systems: Need and development., Indian. J. Pharm. Sci. 2005, 67(3),265 – 272.
View at Publisher | View at Google Scholar - N. R. Jimenez-Castellanos, H. Zia and C. T. Rhodes, “Mucoadhesive drug Delivery Systems”, Drug Dev. Ind. Pharm. 1993, 19, 143.
View at Publisher | View at Google Scholar - D. E. Chickering, J. S. Jacob and E. Mathowitz, “Bioadhesive microspheres II: Characterisation and evaluation of bioadhesion involving hard, bioerodible polymers and soft tissue”, Reactive Polymers, 1995, 25, 189–206.
View at Publisher | View at Google Scholar - V. Iannuccelli, G. Coppi, R. Sansone and G. Ferolla, “Air-compartment Multiple-unit System for Prolonged Gastric Residence, Part II. In vivo Evaluation”, Int. J. Pharma., 1998, 174 (1–2),55– 62.
View at Publisher | View at Google Scholar - R. Wilding and S. P. Newman, “Giving Decisions More Precisions in Drug Development: Use of Radionuclide Imaging,” Drug Dev. Syst. &Scie., 2001, 1 (1) 5–10.
View at Publisher | View at Google Scholar - J. Chen, W. E. Blevins, H. Park and K. Park, “Gastric Retention Properties of Superporous Hydrogel Composites”, J. Control. Rel., 2000, 64 (1–3), 39–51.
View at Publisher | View at Google Scholar - K. Cremer, Drug delivery: Gastro- remaining dosage forms., Pharm.J. 1997, 259, 108.
View at Publisher | View at Google Scholar - Asmussen B, Cremer K, Hoffmann HR, Ludwig K, Roreger M, inventors. Expandable gastroretentive therapeutic system with controlled active substance release in gastrointestinal tract. US patent 6 290 989. September 18, 2001.
View at Publisher | View at Google Scholar - Atyabi F, Sharma HL, Mohammed HAH, Fell JT. In vivo evaluation of a novel gastro retentive formulation based on ion exchange resins. J Control Release. 1996;42:105-113.
View at Publisher | View at Google Scholar - Baumgartner S, Kristel J, Vreer F, Vodopivec P, Zorko B. Optimisation of floating matrix tablets and evaluation of their gastric residence time. Int J Pharm. 2000;195:125-135.
View at Publisher | View at Google Scholar - Bechgaard H, Ladefoged K. Distribution of pellets in gastrointestinal tract. The influence on transit time exerted by the density or diameter of pellets. J Pharm Pharmacol. 1978;30:690-692.
View at Publisher | View at Google Scholar - Bolton S, Desai S, inventors. Floating sustained release therapeutic compositions. US patent 4 814 179. March 21, 1989.
View at Publisher | View at Google Scholar - Bulgarelli E, Forni F, Bernabei MT. Effect of matrix composition and process conditions on casein gelatin beads floating properties. Int J Pharm. 2000;198:157-165.
View at Publisher | View at Google Scholar - Burns SJ, Attwood D, Barnwell SG. Assesment of a dissolution vessel designed for use with floating and erodible dosage forms. Int J Pharm. 1998;160:213-218.
View at Publisher | View at Google Scholar - Chitnis V, Malshe VS, Lalla JK. Bioadhesive polymers synthesis, evaluation and application in controlled release tablets. Drug Dev Ind Pharm. 1991;17:879-892.
View at Publisher | View at Google Scholar - Choi BY, Park HJ, Hwang SJ, Park JB. Preparation of alginate beads for floating drug delivery: effects of CO2 gas forming agents. Int J Pharm. 2002;239:81-91.
View at Publisher | View at Google Scholar - Condtrolled Drug Delivery – Concepts and Advances by S.P.Vyas and Roop K.Khar.
View at Publisher | View at Google Scholar - Dave BS, Amin AF, Patel MM. Gastroretentive drug delivery system of ranitidine hydrochloride: formulation and in vitro evaluation. AAPS PharmSciTech. 2004;5:E34.
View at Publisher | View at Google Scholar - Degtiareva H, Bogdanov A, Kahtib Z, et al. The use of third generation antacid preparations for the treatment of patients with nonulcerous dyspeosia and peptic ulcer complicated by reflux esophagus [in Chinese]. Liakrs’ ka sprava. 1994;5-6:119-122.
View at Publisher | View at Google Scholar - Dennis A, Timminis P, Lel K, inventors. Buoyant controlled release powder formulation. US patent 5 169 638. December 8, 1992.
View at Publisher | View at Google Scholar - Desai S, Bolton S. A floating controlled release drug delivery system: in vitro- in vivo evaluation. Pharm Res. 1993; 10:1321-1325.
View at Publisher | View at Google Scholar - Desai S. A Novel Floating Controlled Release Drug Delivery System Based on a Dried Gel Matrix Network [master’s thesis]. [thesis]. Jamaica, NY: St John’s University; 1984.
View at Publisher | View at Google Scholar - Deshpande AA, Shah NH, Rhodes CT, Malick W. Development of a novel controlled-release system for gastric retention. Pharm Res. 1997;14:815-819.
View at Publisher | View at Google Scholar - El-Kamel AH, Sokar MS, Al Gamal SS, Naggar VF. Preparation and evaluation of ketoprofen floating oral drug delivery system. Int J Pharm. 2001;220:13-21.
View at Publisher | View at Google Scholar - Encyclopedia of Pharmaceutical Technology- vol.6, Edited by James Swarbrick and James C.Boylan.
View at Publisher | View at Google Scholar - Erni W, Held K. The hydrodynamically balanced system: a novel principle of controlled drug release. Eur Neurol. 1987;27:215-275.
View at Publisher | View at Google Scholar - Fabregas JL, Claramunt J, Cucala J, Pous R, Siles A. In vitro testing of an antacid formulation with prolonged gastric residence time (Almagate flot coat). Drug Dev Ind Pharm. 1994; 20:1199-1212.
View at Publisher | View at Google Scholar - Fassihi R, Yang L, inventors. Controlled release drug delivery systems. US patent 5 783 212. July 21, 1998.
View at Publisher | View at Google Scholar - Fix JA, Cargill R, Engle K. Controlled gastric emptying. III. Gastric residence time of a non-disintegrating geometric shape in human volunteers. Pharm Res. 1993;10:1087-1089.
View at Publisher | View at Google Scholar - Floating Drug Delivery Systems: A ReviewArora S, Ali J, Ahuja A, Khar RK, Baboota S. Floating Drug Delivery Systems: A Review. AAPS PharmSciTech. 2005; 06(03):
View at Publisher | View at Google Scholar - Floating drug delivery systems: an approach to oral control drug delivery via gastric retention: Journal of Controlled Release 63(2000) 235-259 Brahma N. Singh, Kwon H. Kim.
View at Publisher | View at Google Scholar - Franz MR, Oth MP, inventors. Sustained release, bilayer buoyant dosage form. US patent 5 232 704. August 3, 1993.
View at Publisher | View at Google Scholar - Garg S, Sharma S. Gastroretentive drug delivery systems. Business Briefing: Pharmatech 2003 Web Site. 5th edition. May 2003
View at Publisher | View at Google Scholar - Gastroretentive drug delivery systems:A ReviewAlexander Streubel. Juergen Stepmann and Roland Bodmeier: Expert Opinion. Drug Deliv.(2006)3(2):217-233
View at Publisher | View at Google Scholar - Gibaly EI. Development and evaluation of novel floating chitosan microcapsules: comparision with non floating chitosan microspheres. Int J Pharm. 2002;249:39-
View at Publisher | View at Google Scholar - Goodman and Gilman’s – The Pharmacological Basis of Therapeutics: 11th Edition; 1243- 1250.
View at Publisher | View at Google Scholar - Groning R, Heun G. Dosage forms with controlled gastrointestinal passage—studies on the absorption of nitrofurantion. Int J Pharm. 1989;56:111-116.
View at Publisher | View at Google Scholar - Groning R, Heun G. Oral dosage forms with controlled gastrointestinal transit. Drug Dev Ind Pharm. 1984;10:527-539.
View at Publisher | View at Google Scholar - Harrigan BM, inventor. Drug delivery device for preventing contact of undissolved drug with the stomach lining. US patent 4 055 178. October 25, 1977.
View at Publisher | View at Google Scholar - Ichikawa M, Watanabe S, Miyake Y, inventors. Granule remaining in stomach. US patent 4 844 905. July 4, 1989.
View at Publisher | View at Google Scholar - Ichikawa M, Watanabe S, Miyake Y. A new multiple unit oral floating dosage system. I: Prepration and in vitro evaluation of floating and sustained-release kinetics. J Pharm Sci. 1991;80:1062-1066.
View at Publisher | View at Google Scholar
