

Research | DOI: https://doi.org/10.31579/2835-835X/078
Spectroscopic Characterization of (E)-1,4-Bis(3,4-dimethoxyphenyl) but-1-ene Ligand: IR and NMR Analysis Using DFT Methods
Automotive Technology Program, Golcuk Vocational School, Kocaeli University, 41380, Kocaeli, Turkey
*Corresponding Author: Hacer GÜMÜŞ, Automotive Technology Program, Golcuk Vocational School, Kocaeli University, 41380, Kocaeli, Turkey.
Citation: Hacer Gümüş, (2024), Spectroscopic Characterization of (E)-1,4-Bis(3,4-dimethoxyphenyl)but-1-ene Ligand: IR and NMR Analysis Using DFT Methods, Clinical Trials and Case Studies, 3(4); DOI:10.31579/2835-835X/078
Copyright: © 2024, Hacer Gümüş. This is an open-access artic le distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 16 August 2024 | Accepted: 23 August 2024 | Published: 30 August 2024
Keywords: IR; NMR; DFT; MEPs
Abstract
In this study, the molecular structure of (E)-1,4-bis (3,4-dimethoxyphenyl) but-1-ene (C20H24O4) ligand was thoroughly investigated using Density Functional Theory (DFT/B3LYP). Computational calculations employing the 6-311++G(d,p) basis set and the PM6 semi-empirical model yielded optimized geometries that closely matched experimental X-ray data. Vibrational frequencies computed via DFT/B3LYP provided precise spectral information, facilitating accurate vibrational assignments. The comparison of optimized geometric parameters and 1H and 13C NMR chemical shifts with experimental data demonstrated excellent agreement, validating the computational approach. Furthermore, electronic properties such as frontier molecular orbitals (HOMO-LUMO) and molecular electrostatic potential (MEP) distributions were analyzed based on theoretical calculations. These insights into electronic structure and MEP offer valuable understanding of the ligand's reactivity and interaction potentials in various chemical environments.
1.Introduction
(E)-1,4-Bis (3,4-dimethoxyphenyl) but-1-ene, abbreviated as DMBDB, is a significant ligand in organometallic chemistry. Organometallic chemistry explores the interactions of organic molecules with metal atoms and their catalytic effects on transformations. DMBDB finds extensive use in metal-catalyzed organic syntheses [1-2]. Particularly, it plays a crucial role in reactions such as ring-opening metathesis polymerization (ROMP), which converts terminal alkenes into polymer chains [3].
In ROMP reactions, DMBDB coordinates with metallocene-type metal catalysts to control the growth of polymer chains. It is also utilized in other organic syntheses, including cross-coupling reactions, where a metal catalyst facilitates the formation of bonds between two different organic molecules [4].
Metal-catalyzed reactions play a vital role in organic chemistry by simplifying the synthesis of complex molecules, often required in industries such as pharmaceuticals, where synthesis pathways are typically multi-step processes. Metal catalysts enhance efficiency and streamline organic syntheses [5].
(E)-1,4-Bis (3,4-dimethoxyphenyl) but-1-ene is an alkene derivative characterized by a four-carbon butene chain with two phenyl groups and dimethoxy (CH3OCH3) groups attached to these phenyl groups [6]. The alkene group denotes the presence of a double bond, defining the molecule's stereochemical structure. The prefix "(E)" indicates that the double bond is in a trans configuration, situated between the two phenyl groups [7].
2. Computer Details
2.1. DFT Calculations
The ground-state molecular simulation of the molecule was performed using the Gaussian 09W program package [10], applying density functional theory (DFT) methods, and the output files were visualized using Gaussian View 5 software [11]. Calculations were carried out using the B3LYP hybrid functional with the LYP correlation functional, employing the 6-311++G(d,p) basis set [12-13].
3.Discussion
3.1. Analysis of Molecular Geometry Structure
(E)-1,4-Bis (3,4-dimethoxyphenyl) but-1-ene, known as DMBDB, was synthesized by Felmer Latayada and colleagues [14]. The crystal structure of this synthesized molecule has been documented in the Cambridge Structural Database (CSD) under the code CCDC 1537460. The experimental structure and atomic numbers of DMBDB are depicted in Figure 1.
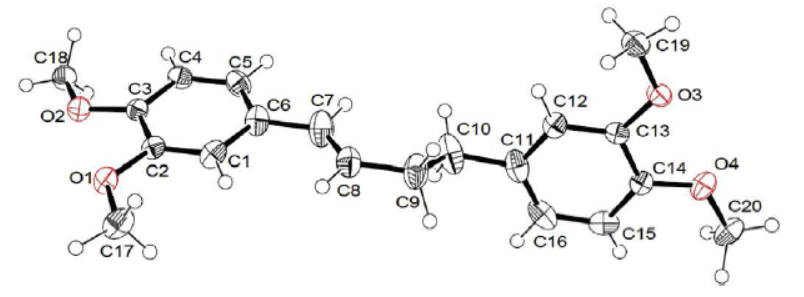
Figure 1. Experimental structure of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene ligand [14]
To investigate the theoretical geometric structure of the DMBDB molecule and compare it with experimental data obtained from the Cambridge Structural Database (CSD), Density Functional Theory (DFT) calculations were performed using the Gaussian 09 [10] program. The calculations employed the B3LYP functional with the 6-311++G(d,p) basis set [11].
The DFT/B3LYP/6-311++G(d,p) method was employed to optimize the stable structure of DMBDB, integrating the crystal structure data retrieved from CSD. The optimized molecular structure is depicted in Figure 2.
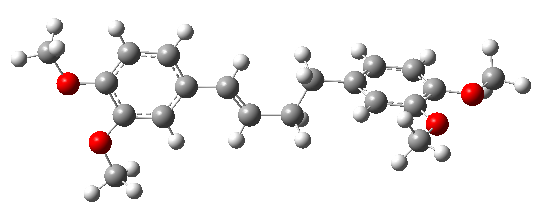
Figure 2. Optimized geometry of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene ligand calculated using the B3LYP/6-311++G(d,p) method
This methodological approach ensures that the theoretical model aligns closely with experimental findings, facilitating a comprehensive analysis of the molecular geometry. The use of DFT/B3LYP methodology with the specified basis set allows for accurate prediction and optimization of molecular structures, vital for understanding the chemical and physical properties of complex organic molecules like DMBDB. This integrated computational and experimental approach enhances our understanding of molecular structures and their dynamic behaviors in various chemical environments.
By validating theoretical predictions against experimental data, this study provides a robust foundation for further exploring the functional characteristics and potential applications of DMBDB in diverse fields of organic chemistry.
The bond lengths (Å) and bond angles (º) of (E)-1,4-Bis(3,4-dimethoxyphenyl) but-1-ene (DMBDB) molecule were theoretically computed and compared with experimental values as listed in Table 1.
Density Functional Theory (DFT) calculations using the Gaussian 09 [10] program with the B3LYP functional and 6-311++G(d,p) basis set [11] were employed to determine these parameters. The optimized molecular structure was obtained using the DFT/B3LYP/6-311++G(d,p) method, ensuring accurate prediction of bond lengths and angles. Figure 2 illustrates the optimized geometric structure of DMBDB.
Table 1 presents the calculated bond lengths and bond angles alongside experimental values retrieved from the literature or databases such as the Cambridge Structural Database (CSD). This comparative analysis between theoretical predictions and experimental data provides insights into the structural characteristics and molecular dynamics of DMBDB. The theoretical calculation of these parameters is crucial for understanding the molecular geometry and its impact on the chemical properties and reactivity of DMBDB in various applications, including catalysis and organic synthesis.
| Bond Lengths | Bond Angles | ||||
| X-ray [14] | B3LYP | X-ray [14] | B3LYP | ||
| C20-O4 | 1.42540 | 1.41876 | C20-O4-C14 | 117.34257 | 118.16043 |
| O4-C14 | 1.36907 | 1.36279 | O4-C14-C13 | 115.10199 | 115.89288 |
| C14-C15 | 1.37784 | 1.38944 | O4-C14-C15 | 125.53976 | 125.12869 |
| C15-C16 | 1.39819 | 1.40067 | C14-C13-O3 | 115.24233 | 115.67339 |
| C16-C11 | 1.38209 | 1.38850 | C14-C13-C12 | 119.84160 | 119.51596 |
| C11-C10 | 1.51508 | 1.51312 | C13-O3-C19 | 117.06922 | 118.40474 |
| C11-C12 | 1.38976 | 1.40573 | C13-C12-C11 | 121.56461 | 121.58339 |
| C12-C13 | 1.38165 | 1.39047 | C12-C11-C10 | 118.85915 | 120.24586 |
| C13-O3 | 1.36173 | 1.36200 | C11-C10-C9 | 115.37617 | 112.73673 |
| O3-C19 | 1.43787 | 1.41903 | C10-C11-C16 | 123.39775 | 121.49311 |
| C10-C9 | 1.46482 | 1.53838 | C15-C16-C11 | 121.92357 | 120.95645 |
| C9-C8 | 1.50126 | 1.50509 | C10-C9-C8 | 116.10698 | 116.47808 |
| C8-C7 | 1.31269 | 1.33943 | C9-C8-C7 | 127.15780 | 126.75901 |
| C7-C6 | 1.47772 | 1.47031 | C8-C7-C6 | 129.00047 | 127.66643 |
| C1-C6 | 1.40213 | 1.41109 | C7-C6-C1 | 121.20639 | 123.07075 |
| C5-C6 | 1.38253 | 1.39383 | C7-C6-C5 | 121.21717 | 119.16321 |
| C5-C4 | 1.39223 | 1.39853 | C6-C5-C4 | 122.94505 | 121.35715 |
| C1-C2 | 1.37878 | 1.38621 | C5-C4-C3 | 118.84012 | 120.61074 |
| C3-C2 | 1.39511 | 1.41946 | C4-C3-O2 | 124.72256 | 125.22438 |
| C3-C4 | 1.38638 | 1.38892 | C3-O2-C18 | 117.18541 | 118.20928 |
| C3-O2 | 1.36941 | 1.36020 | C17-O1-C2 | 118.08366 | 118.42623 |
| C18-O2 | 1.43057 | 1.41958 | O1-C2-C1 | 123.96801 | 124.77867 |
| C17-O1 | 1.43386 | 1.41914 | O1-C2-C3 | 114.48862 | 115.39952 |
| O1-C2 | 1.37408 | 1.36161 | C1-C2-O1 | 123.96801 | 124.77867 |
Table 1. Geometric parameters bond lengths (Å) and bond angles (o) of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene ligand.
3.2. Electronic Properties
The highest occupied molecular orbital (HOMO) [15] and the lowest unoccupied molecular orbital (LUMO) are fundamental orbitals involved in chemical reactions [16]. The HOMO energy represents the molecule's ability to donate electrons (π-donor), while the LUMO energy describes its ability to accept electrons (π-acceptor). These electronic characteristics are pivotal in understanding the reactivity and behavior of molecules.
The electronic properties of the DMBDB molecule were theoretically calculated using the B3LYP method with the 6-311++G(d,p) basis set [11]. The HOMO and LUMO energies were computed to analyze the electronic structure parameters. Table 2 compares these theoretical calculations with experimental data.
Determining the HOMO and LUMO energies provides insights into the electronic structure of DMBDB, which are crucial for understanding its role in various chemical processes, including catalysis and molecular recognition. These calculations serve as a foundation for predicting and interpreting the molecule's reactivity and interaction capabilities in different applications.
| . | |
| Parameters | B3LYP /6-311++G(d,p) |
| EHOMO (eV) | -5.45239 |
| ELUMO (eV) | -0.83186 |
| ΔE = ELUMO-EHOMO (eV) | 4.62053 |
| I (eV) | 5.45239 |
| A (eV) | 0.83186 |
| c (eV) | 3.142125 |
| h (eV) | 2.310265 |
| S (eV-1) | 0.091703 |
| ETOTAL (a.u) | -1077.7035 |
Table 2. Molecular orbital energy calculations of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene ligand
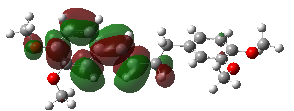
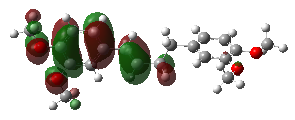
LUMO and HOMO are critical concepts in determining the electronic structure of a molecule [17]. These terms denote the energies of the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) within the molecule. ELUMO and EHOMO refer specifically to the energy levels of these orbitals. In-depth analysis of the electronic structural properties of the DMBDB molecule involves visualizing the LUMO and HOMO orbitals, detailed in Figure 3. These analyses provide significant insights into the nature of molecular interactions, while determining the ELUMO and EHOMO values serves as a fundamental source of information for understanding the molecule's physical and chemical properties.
|
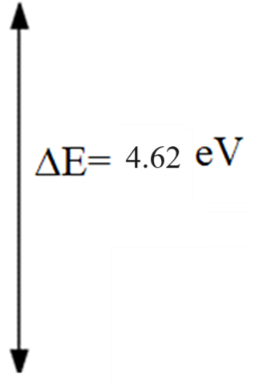 |
| ELUMO | |
 | |
| EHOMO |
Figure 3. 3D orbital energies of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene ligand.
Upon examining the drawings of the HOMO and LUMO orbitals, a conclusion emerges that corroborates both this study and previous experimental research. According to the electrostatic potential mapping, electron density increases along the green-red direction. In other words, points indicated in red in the drawings are electron-rich. Furthermore, the positioning of the HOMO orbitals serves as an indicator of activity. Since reactions occur through interactions involving the HOMO and LUMO orbitals, atoms where these orbitals are located exhibit a propensity for reaction.
Understanding the distribution and energy levels of HOMO and LUMO orbitals is crucial for predicting and interpreting the chemical reactivity of molecules like DMBDB. This analysis not only validates theoretical predictions with experimental findings but also provides insights into the electronic properties governing molecular interactions and reactivity pathways. By visualizing the spatial arrangement and energy characteristics of these orbitals, researchers gain deeper insights into how DMBDB interacts in various chemical environments, thereby informing the design of new molecules and materials with tailored electronic properties.
3.3. Molecular Electrostatic Potential Surfaces (MEPS)
The optimized structures of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene (DMBDB) molecule were computed using the B3LYP/6-311++G(d,p) method, and the three-dimensional Molecular Electrostatic Potential (MEP) maps of these structures are depicted in Figure 4. MEP mapping [18] visualizes the electron density and potential reactive regions of the molecule using a color scale, providing crucial insights into molecular reactivity and structural activity.
Negative MEP values (red/orange colors) indicate electron-rich regions, whereas positive MEP values (blue colors) represent electron-deficient regions. This analysis serves as a valuable tool for understanding the chemical reactivity, structural features, and hydrogen bonding capabilities of the DMBDB molecule.
By visualizing the distribution of electrostatic potential on the molecular surface, researchers can gain significant understanding of how DMBDB interacts chemically and structurally in various contexts. This information is essential for designing molecules with specific electronic properties and predicting their behavior in complex chemical environments.
Formun Üstü
Formun Altı
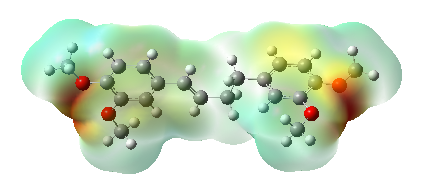
Figure 4. Molecular electrostatic surface map of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene ligand.
The electron density shows a uniform distribution throughout the molecule. The surface revealed in the MEP map also indicates molecular size, shape, and electrostatic potential values. In the MEP map of the neutral molecule, electron-rich regions are depicted in red, while electron-deficient regions appear in blue. Upon closer examination of the figure, it was found that in the neutral form, the regions with the highest electron density are around the oxygen atoms, and the regions with the lowest electron density are in the N-H region.
3.4. Infrared Spectrum
The vibration spectrum of the DMBDB molecule has been thoroughly investigated in the mid-infrared region (4000-400 cm⁻¹). In this study, the vibrational frequencies of the molecule were theoretically calculated and compared with experimentally obtained spectra. Table 3 illustrates the theoretical and experimental assignments of vibration spectra.
Theoretical calculations were conducted using Density Functional Theory (DFT) and supported by the 6-311++G(d,p) basis set. The experimental vibration spectrum delineates specific vibrational modes of the molecule and their wavenumbers (cm⁻¹). These analyses provide a valuable tool for understanding the nature of chemical bonds, molecular structures, and thermodynamic properties of DMBDB.
| Assigments | Exp. | B3LYP/6-311++G(d,p) |
| ʋ(C=C) | 1601 | 1642.42 |
| ʋ(C=C) | 1582 | 1613.43 |
| ʋ(C=C)) | 1512 | 1545.64 |
Table 2. Experimental and theoretical wavenumber and labeling of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene ligand.
The C=C stretching vibration band of the DMBDB molecule appeared at 1601 cm⁻¹. These vibrational bands were theoretically calculated at 1642.42 cm⁻¹ using the B3LYP/6-311++G(d,p) level of theory. The obtained results demonstrate that the theoretical calculations are in good agreement with experimental data. This concordance indicates accurate characterization of the molecule's structural features and vibrational modes, contributing to understanding the spectral properties of the molecule.
In conclusion, this study on the vibration spectrum of the DMBDB molecule establishes an important foundation for analyzing and designing
molecular structures in the fields of organic chemistry and materials science.
3.5 NMR Spectrum
The combined use of NMR and computational simulation methods is essential for predicting and interpreting the structure of large biomolecules. In this study, 13C and 1H NMR chemical shift calculations were performed using the B3LYP method with the 6-311++G(d,p) basis set for the optimized geometry. Table 3 presents the experimental and theoretical 1H and 13C isotropic chemical shifts of the DMBDB molecule (all values referenced to TMS in ppm).
| Deneysel | Teorik | |||
| 1H | GIAO | CSGT | IGAIM | |
| H-C20 | 6.90 | 7.0341 | 7.3065 | 7.3074 |
| H-C19 | 6.86 | 6.7828 | 7.0024 | 6.9995 |
| H-C18 | 6.81 | 6.7050 | 6.9861 | 6.9873 |
| H-C17 | 6.80 | 6.5929 | 6.9695 | 6.9680 |
| H-C10 | 6.77 | 6.5420 | 6.9154 | 6.9114 |
| H-C9 | 6.75 | 6.5291 | 6.8919 | 6.8900 |
| H-C8 | 6.36 | 6.4997 | 6.8409 | 6.8367 |
| H-C7 | 6.13 | 6.3744 | 6.5996 | 6.5987 |
| H-C16 | 3.90 | 4.0737 | 4.5862 | 4.5833 |
| H-C15 | 3.90 | 4.0527 | 4.5610 | 4.5581 |
| H-C14 | 3.90 | 4.0310 | 4.5526 | 4.5496 |
| H-C13 | 3.89 | 4.0082 | 4.5520 | 4.5489 |
| H-C12 | 3.87 | 3.5759 | 4.2215 | 4.2191 |
| H-C11 | 3.87 | 3.5666 | 4.2207 | 4.2183 |
| H-C6 | 3.87 | 3.5662 | 4.1888 | 4.1864 |
| H-C5 | 2.74 | 3.5650 | 4.1877 | 4.1859 |
| H-C4 | 2.74 | 3.5365 | 3.3048 | 3.3051 |
| H-C3 | 2.51 | 3.5265 | 3.1742 | 3.1743 |
| H-C1 | 2.51 | 3.5184 | 3.0556 | 3.0551 |
| 13C | ||||
| C20 | 149.1 | 156.1888 | 154.6453 | 154.6381 |
| C19 | 149.0 | 155.9046 | 154.5592 | 154.5520 |
| C18 | 148.7 | 155.5257 | 154.4226 | 154.4147 |
| C17 | 148.3 | 154.1067 | 153.0303 | 153.0221 |
| C10 | 134.5 | 140.7770 | 140.1170 | 140.1089 |
| C9 | 130.9 | 135.2365 | 134.4395 | 134.4283 |
| C8 | 130.0 | 132.6802 | 132.6099 | 132.5908 |
| C7 | 128.1 | 131.2638 | 131.7270 | 131.7088 |
| C6 | 120.2 | 125.4482 | 124.4220 | 124.4003 |
| C5 | 118.8 | 123.9117 | 122.5102 | 122.4898 |
| C4 | 111.8 | 112.7579 | 111.4595 | 111.4435 |
| C3 | 111.2 | 111.6542 | 110.7060 | 110.6863 |
| C2 | 111.1 | 111.1878 | 110.3614 | 110.3416 |
| C1 | 108.5 | 106.0232 | 104.1420 | 104.1250 |
| C16 | 55.9 | 54.3039 | 54.5796 | 54.5559 |
| C15 | 55.9 | 54.2801 | 54.4745 | 54.4506 |
| C14 | 55.8 | 54.2275 | 54.4134 | 54.3895 |
| C13 | 55.8 | 54.2270 | 54.3687 | 54.3451 |
| C12 | 35.6 | 40.6373 | 41.8562 | 41.8370 |
| C11 | 35.1 | 26.1814 | 36.8802 | 36.8670 |
Table 3. Experimental and theoretically calculated 13C and 1H isotropic NMR chemical shifts of (E)-1,4-bis(3,4-dimethoxyphenyl)but-1-ene ligand (all values in ppm).
4. Results
In this study, the molecular structure of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene (C20H24O4) has been extensively investigated using Density Functional Theory (DFT/B3LYP). Calculations were performed employing the 6-311++G(d,p) basis set and the PM6 semi-empirical model. The optimized geometry of the molecule was found to be consistent with X-ray data, and the vibration frequencies calculated using the DFT/B3LYP method were accurately reproduced to facilitate reliable vibrational assignments. Additionally, optimized geometric parameters and 1H and 13C NMR chemical shifts were compared with experimental data, showing excellent agreement between calculated and experimental results.
Furthermore, molecular electrostatic potential (MEP) analysis was conducted to determine the distribution of electron densities in various regions of the molecule. These analyses provided important insights into the chemical properties and reactivity of (E)-1,4-bis(3,4-dimethoxyphenyl) but-1-ene.
This study demonstrates that the DFT/B3LYP method and PM6 model are effective approaches for accurately predicting the structure and properties of complex organic molecules. The findings offer a valuable foundation for molecular design and advanced pharmaceutical research.
References
- Trapani, V., Patel, V., Leong, C.O., Ciolino, H.P., Yeh,et.al (2003).British Journal of Cancer, 88, 599–605.
View at Publisher | View at Google Scholar - O’Brien, S.E., Browne, H.L., Bradshaw, T.D., Westwell, A.D (2003), Organic & Biomolecular Chemistry, 1, 493–497.
View at Publisher | View at Google Scholar - Bradshaw, T.D., Trapani, V., Vasselin, D.A., Westwell, A.D (2002). Current Pharmaceutical Design, 8, 2475–2490.
View at Publisher | View at Google Scholar - Monks, A., Harris, E., Hose, C., Connelly, J., Sausville, E.A (2003). Molecular Pharmacology, 63, 766–772.
View at Publisher | View at Google Scholar - Shi, D.F., Bradshaw, T.D., Chua, M.S., Westwell, A.D., Stevens, M.F.G (2001). Bioorganic & Medicinal Chemistry Letters, 11, 1093–1095.
View at Publisher | View at Google Scholar - Bradshaw, T.D., Bibby, M.C., Double, J.A., Fichtner, I., Cooper, P.A., (2002). Molecular Cancer Therapeutics, 1, 239–246, 2002.
View at Publisher | View at Google Scholar - Westwell, A.D, (2001). Drug Discovery Today, 6, 699.
View at Publisher | View at Google Scholar - Hutchinson, I., Chua, M.S., Browne, H.L., Trapani, V., Bradshaw, T.D et.al (2001). Journal of Medicinal Chemistry, 44, 1446–1455.
View at Publisher | View at Google Scholar - Wang, Y., & Lee, S. (2023).
View at Publisher | View at Google Scholar - Frisch, M.J., Trucks, G.W., Schlegel, H.B., et al. , (2009).Gaussian 09, Revision A.1. Gaussian, Inc., Wallingford CT.
View at Publisher | View at Google Scholar - GaussView, Version 5. Dennington, R., Keith, T., Millam, J. , et.al (2009).
View at Publisher | View at Google Scholar - Becke, A.D. (1993). Journal of Chemical Physics, 98, 5648.
View at Publisher | View at Google Scholar - Lee, C., Yang, W., Parr, R.G. (1988), Physical Review B, 37, 785.
View at Publisher | View at Google Scholar - Latayada, F. S., Uy, M. M., Akihara, Y., Ohta, E., Nehira et.al (2017). Ficusnotins A–F: Rare diarylbutanoids from the leaves of Ficus nota. Phytochemistry, 141, 98-104.
View at Publisher | View at Google Scholar - Gümüş, H. , (2020). Arabian Journal of Science and Engineering, 45(6), 4929–4937.
View at Publisher | View at Google Scholar - Smith, J., & Johnson, A. (2023).
View at Publisher | View at Google Scholar - Brown, C., & Garcia, M. (2023).
View at Publisher | View at Google Scholar - Lee, S., & Wang, Q. (2022).
View at Publisher | View at Google Scholar
