

Research Article | DOI: https://doi.org/10.31579/2835-8376/039
Possible Association of a Novel C.7664a>C Variant of Celsr1 with Primary Lymphedema: Family Case Report
- Y.V. Filina *
- A.K. Feiskhanov 1
- ,A.R. Ibragimova 2
- E.A. Boulygina 1
- M.N. Zhuravleva
- A.A. Rizvanov 1
- R.R. Miftakhova 1
1 Kazan Federal University.
2 Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences.
*Corresponding Author: Y.V. Filina, Kazan Federal University
Citation: Y.V. Filina , A.K. Feiskhanov , A.R. Ibragimova , E.A. Boulygina , M.N. Zhuravleva , A.A. Rizvanov and R.R. Miftakhova (2025). Title: Skin tuberculosis among Sudanese multi drugs resistance patients at Abu Anja teaching hospital, Clinical Research and Reviews.4(1); DOI: 10.31579/2835-8376/039.
Copyright: © 2025, Y.V. Filina, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 02 January 2025 | Accepted: 15 January 2025 | Published: 29 January 2025
Keywords: lymphedema; celsr1; exome sequencing; family case
Abstract
Lymphedema refers to a group of disorders characterized by the accumulation of protein-rich fluid in the interstitial tissues, resulting from an imbalance between lymph production and it’s drainage via lymphatic vessels. More than 10 types of isolated primary LD is described, and among them CELSR1-associated lymphedema are one of the less studied. A family with two sibs, a 17-year-old boy and an 8-year-old girl, both diagnosed with primary LD, presented to the clinic. Both patients were observed with primary symptoms of LD at 2-3 months of age, presented by the swelling of right foot. Lymphatic pathologies were not diagnosed in parents or close relatives. The rare c.7664A>C variant, resulting in the H2555P missense mutation in the intracellular loop of CELSR1 transmembrane domain, was discovered in a patient. The clinical signs, MRL scanning data, in silico predictors and the absence of known pathogenic mutations or rare variants of unknown significance in other genes, associated with the development of primary lymphedema, allow us to assume that c.7664A>C variant of CELSR1 may be responsible for pathogenesis of the disease in the presented family case.
1.Introduction:
Primary lymphedema (PLD), including isolated, syndromic, and systemic forms, occurs at a frequency of approximately 1-1.15 per 100,000 individuals under the age of 20 [1,2]. PLD arises from congenital anomalies and is associated with dysplasia, hypoplasia, or hyperplasia of components of the lymphatic system, caused by genetic mutations. In 1998, the first association between mutations in the FLT4 (VEGFR3) gene and primary congenital lymphedema was demonstrated, followed by the discovery that lymphedema-distichiasis can be caused by FOXC2 mutations in 2000 [3]. Mutations in seven genes, FLT4, GJC2, FOXC2, SOX18, GATA2, CCBE1, and PTPN14, were shown to be responsible for the development of 36% of inherited and 8% of sporadic lymphedema cases[4]. Most of them are associated with the VEGFR3 signaling pathway, the key player of embryonic lymph angiogenesis. There are increasing evidence of an association between pathways involved in vascular morphology and the development of PLD. The examples include the adhesion receptor CELSR1 and lymphatic malformation 9 (OMIM: 619319), atypical cadherin FAT4 and Hennekam lymphangiectasia-lymphedema syndrome 2 (OMIM: 616006), connexin GJC2 and lymphatic malformation 3 (OMIM: 613480), and the mechanosensitive ion channel component PIEZO1 and lymphatic malformation 6 (OMIM: 616843) [5].
2.Materials And Methods:
Patient’s recruitment
Patients were treated at the Lymphology Center of the Kazan Federal University in 2021-2023. Study was approved by the local ethical committee (2020-12-28, #27) of the Kazan Federal University. Written informed consent was obtained for all patients.
MRI-Magnetic resonance lymphography (MRL) was performed using a 3.0 T MR Siemens Magnetom Verio instrument. 3D-TSE heavily T2-weighted magnetic resonance imaging was performed, data were analyzed by Syngo 3D for MIP transformation (Siemens Medical Solutions, USA).
Exome Sequencing and Analysis
DNA was isolated from blood samples using the QIAAmp DNA mini kit (Qiagen, USA) according to the manufacturer's instructions. DNA libraries were prepared using Agilent SureSelectXT Human All Exon v8 (Agilent Technologies, USA) and sequenced on the NextSeq 500 platform (Illumina Inc., USA). Raw reads were mapped to the human reference genome hg38 using BWA v. 0.7.17 [6]. The improvement of reads alignment and whole exome variant calling were performed using the Genome Analysis Tool Kit v.4 [7] according to the GATK Best Practices recommendations [8]. For single nucleotide variant (SNV) calling, the Haplotype Caller module was used. High-quality SNVs were filtered using vcf-annotate [9] with the following parameters: MinDP=20, Qual=10, and MinMQ=10, and annotated using wANNOVAR [10]. Synonymous variants and variants with MAF ≥1?cording to the GnomAD (v. 3.1.2, total) [11] and RUSeq [12] databases were excluded from variant analysis. PolyPhen2, SIFT, MutationTaster, and CADD algorithms were used to predict the contribution of the detected variants to the functional activity of CELSR1. Clinical significance was assessed according to the ACMG criteria [13].
Sanger Sequencing
Direct sequencing of exon 25 of CELSR1 were performed using ABI 3730 DNA Analyzer (Thermo Fisher Scientific, USA). Primers for PCR and sequencing were designed with PrimerQuest Tool (Integrated DNA Technologies, USA) and synthesized by Genera, LLC (Russia). Primer’s sequences were the follows (5’→3’):
Forward: GGGACTGGGTTTGTGTTCAT Reverse: GGTCTGTTGGAAATCTGTGGT
CELSR1 target region was amplified by Q5 DNA polymerase (NEB, USA) according to the manufacturer’s guide with Ta=65°C.
3.Results:
3.1.Family Case
Children were born in a family of non-consanguineous parents; lymphatic pathologies were not diagnosed in parents or closer relatives in 3 generations. Mother was diagnosed with chronic pyelonephritis, toxoplasmosis, herpes simplex virus, and cytomegalovirus infection. Patient’s aunt (mother’s sister) has frequent recurrent erysipelas without visual signs of foot edema. Male patient was delivered by natural vaginal childbirth as the first child of a first gravida with birth weight of 3454 g and Apgar score 8/9. Intrauterine left-sided hydronephrosis was diagnosed using routine antenatal ultrasound examination. Surgical treatment of vesicoureteral reflux was performed after birth, and secondary shrinkage of the left kidney and chronic pyelonephritis developed. At two months increased volume of the right foot was observed, at three months lymphostasis was confirmed by medical examination. Complex Physical Decongestive Therapy (CPDT) was carried out irregularly from the age of 11; positive result was achieved after 4 courses of therapy at 13 years, whereas intradermal thrombosis of the right foot was diagnosed at 15. At the age 17 patient had II-III stage lymphedema of right lower limb, including foot and lower third of the leg, with recurrent erysipelas, lymphocysts, papillomas. After CPDT symptoms decreased significantly (Figure 1 A-C).
Figure 1: Photographs of foot of male patients (A-B), male patient after thrombosis (C) and female patient (D). His younger sister
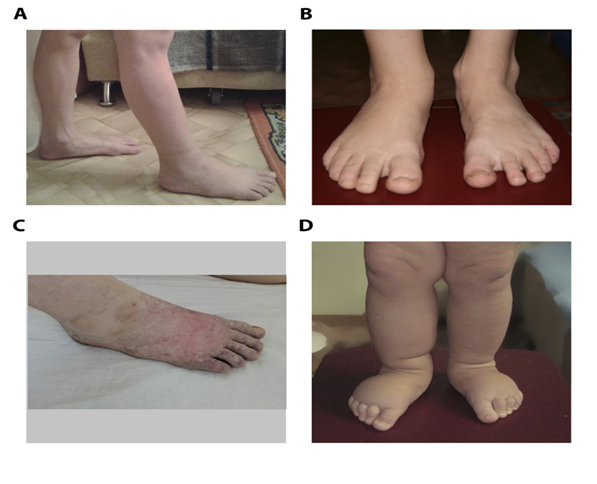
was delivered by natural vaginal childbirth as the second child of a second gravida with birth weight of 3994 g and Apgar score 8/9. Mixed CNS lesion, congenital adrenal hyperplasia and virilization was diagnosed after birth, whether her karyotype test showed no deviations. Prenatal lesions associated with lymphedema or other disorders were not discovered, edema of right lower limb was detected at 2-3 months, left lower limb edema was developed at 10 months (Figure 1D). Patient underwent treatment with CPFT 1-2 times a year with positive results from 2 years, and at the age 8 she had II stage lymphedema of both lower limbs, including foot and leg, without skin lesions.
3.2.Family Case
Both patients were examined by MRL without contrast agent. No pathogenic changes were revealed in the thoracic duct, whereas hypoplastic and aplastic lymphatic components in lower extremities were discovered (Figure 2).

Figure 2: MRL images of male (A) and female (B) patients indicating reduction of lymphatic vasculature (white arrows), subcutaneous thickness (yellow arrows) and edema (blue arrows). In boy on the right thigh in the upper third of the lower extremities, the femoral lymphatic vessel was not visualized, proximally and distally on the thigh vessels were not changed, deep lymphatic vessels of the muscles in the right leg were 2 times smaller than in the left. The medial and lateral subcutaneous lymphatic collectors of the right lower limb were not visualized, and subcutaneous lymphatic vessels were represented as diffuse small vessels. In girl hypoplastic deep lymphatic vessels of the lower legs and thighs were visualized. The medial and lateral subcutaneous lymphatic collectors of both lower extremities were aplastic and not visualized. The subcutaneous lymphatic vessels were represented as diffuse small vessels.
3.3.Exome Analysis
Exome variants were sorted by the frequency according to GnomAD 3.1 database and those with frequency ≤ 1% were selected for further analysis. 467 common variants in 360 genes were identified, and among them only CELSR1 was shown to be associated with PLD. No pathogenic or even rare variants with uncertain/unknown significance were found in the 87 other genes associated with isolated or syndromic PLD, including FLT4, GJC2, FOXC2, SOX18, GATA2, CCBE1, and PTPN14, which have been linked with the most cases of the disease [4]. Genomic c.7664A>C variant was identified as a joint variant for two siblings. Variant was additionally analyzed by Sanger sequencing in children and their parents. Heterozygous change was confirmed in both children and their mother, whereas father was unaffected (Figure 3).
Figure 3. The family tree showing the c.7664A>C carriers without swellings (gray), LD affected (black), and unaffected members with wild type CELSR1 (white). In silico predictors SIFT, Plyphen2 and Mutation tester, as well as the evolutionary conservation assessment instruments alone, were used to estimate the possible influence of c.7664A>C variant on the protein function, and all of them predicted this variant to be deleterious (Table 1).
Table 1: Characterization of the c.7664A>C (p.H2555P) variant by in silico prediction tools.
Variant | Title 2 |
|---|---|
Exon | 25 |
Protein domain | Transmembrane |
SIFT prediction | Deleterious (D) |
PolyPhen-2 prediction | Probably damaging (D) |
Mutation Taster prediction | Disease causing (D) |
100 vertebrates conservation by PhastCons | 1.0 |
4.Discussion:
CELSR, planar cell polarity organizer, forms trans-dimers and is involved in the organization of polarized structures between neighboring cells [14,15] and have a coordinating effect in the propagation of a signal from cell to cell [16,17]. Animal models revealed, that these process are necessary for the proper formation of the neural tube and its derivatives[18,19], stereocilia and neurons in the inner ear[20,21], ciliated cells of the fallopian tube[22] and other tubular organs, hair[23], and endothelial valve cells[24]. Nevertheless, the functioning of CELSR family proteins at the molecular level remains largely unclear. Like most adhesion GPCRs, they are orphan receptors with an unknown activation mechanism. Even the assumption of a connection between CELSR and heterotrimeric G-protein is based only on the presence of a 7-TM domain characteristic of GPCRs, and the site that interacts with any G-protein has not yet been identified. Experimental data indicate that adhesion G protein–coupled receptors (ADGRs) can activate signaling events unrelated to G- proteins but based on homophilic trans-interactions necessary for planar cell polarity[15]. Disruptions in CELSR1 in humans are associated with the development of lymphatic malformation 9 (LM9, OMIM: 619319). The expression of Celsr1 in mouse vessels begins during the formation of valves at embryonic day 16 (E16-E16.5) [24], which is approximately equivalent to 23 weeks of gestation in humans [25]. During this process, endothelial cells elongate, undergo reorientation, and coordinated migration into the lumen of the vessel, where they initiate valve leafletformation. Knockout of Celsr1 in mice leads to disruption of intercellular contacts, endothelial cells are unable to rearrange and adopt a perpendicular orientation for valve formation, leading to aplasia and impeding the ability of the lymphatic system to collect and drain fluid from tissues, ultimately resulting in lymphedema[26]. However, Celsr1 is necessary for development but not for maintaining the structure of lymphatic vessels - knockouts after valve formation do not have a significant impact on vessel structure[24]. CELSR proteins are non-classical cadherins that do not interact with catenins, unlike classical representatives. The structure of CELSRs includes a large ectodomain, with 8 or 9 N-terminal cadherin repeats, up to 8 epidermal growth factor-like domains, two laminin G-type repeats, an EGF- like laminin, one hormone receptor motif (HRM), and a GPCR Autoproteolysis Induction (GAIN) domain. The membrane part consists of a 7-transmembrane domain (7-TM), and the intracellular cytoplasmic tail has 300-600 amino acid residues [15]. The GPS region in the GAIN domain of most ADGRs is responsible for autoproteolysis in the extracellular domain, which occurs in the ER during receptor biosynthesis. Mature ADGRs are usually cleaved and exist as a non-covalently bound complex of N- and C-terminal fragments, but some of them, including CELSR1, do not cleave due to the absence of a consensus catalytic sequence (Leu↓Thr/Ser/Cys) in GPS [15]. Cadherin-like, EGF-like, and laminin-like repeats confer adhesive properties and facilitate intercellular communication, while GAIN and laminin domains can bind unidentified ligands. Transmembrane domain of CELSR1 is relatively small (~8% of all amino acids), and contains few known mutations, especially those associated with diseases. We discovered rare variant, c.7664A>C, resulted in the substitution of histidine to proline at position 2555 (H2555P) of the intracellular loop in the CELSR1 transmembrane domain in a brother and sister with lower extremity lymphedema caused by aplasia and hypoplasia of lymphatic structures. The oedema in both children appeared in the first year of life, and LD was not diagnosed in of other family members. Despite the low/unknown frequency of the detected polymorphism in the population, which is one of the pieces of evidence of pathogenicity [13], identified variant cannot be confidently classified as pathogenic or neutral. The possible pathogenicity of H2555P, in addition to its frequency of occurrence, is evidenced by its presence in two close relatives with primary lymphedema and by the results of in silico analysis predicting a damaging/negative effect of the mutation on protein function. However, no missense mutation in the transmembrane domain of CELSR1 has been previously classified as pathogenic in LM, and no functional analysis of such alterations has been performed. The pathogenic/likely pathogenic findings closest to p.H2555 include p.A2510P/V and p.D2601N, revealed in the patients with congenital heart defects and primary angle closure glaucoma [27–29]. To date, less than 10 experimental studies have been published describing the genetic features and clinical-phenotypic characteristics of CELSR1-associated lymphedema (Table 2). It has been shown that the disease belongs to forms with early manifestation (first decade of life), characterized by incomplete penetrance and variable expression, predominantly affecting females.
DNA change | Protein change | Main features | Reference |
|---|---|---|---|
c.5871G>A | p.Trp1957* | Inactivating variant of CELSR1 identified in 5 family members with confirmed lower limb lymphedema; absent in 6 healthy individuals. Proband had leg swelling at 10 years, tortuous vessels, extensive retrograde lymph flow, and unique "sheet flow" in both legs. | [30] |
c.5121dupC | p.Ile1708fs*44 | Inactivating mutation of CELSR1 in two patients with lymphatic disorders and their asymptomatic father. Lymphoscintigraphy showed lymphatic abnormalities correlated with disease burden. Carriers included 5 women and 4 men; only women showed clinical manifestations. | [31] |
c.5526+2T>A | NA | 5 individuals with inactivating CELSR1 variants | [32] |
c.6739+1G>A | NA | Identified in 95 probands with primary lymphedema. Four women had childhood/adolescence onset with unilateral/bilateral lower limb edema. A man with c.5702-1G>C developed disease at 77; family history showed lymphedema in 83% of women and 25% of men with mutations. | [32] |
c.868G>T | p.Glu290* | Identified in 95 probands with primary lymphedema (see above) | [32] |
c.2042del | p.Asn681Metfs*16 | Identified in 95 probands with primary lymphedema (see above) | [32] |
c.5702-1G>C | NA | Identified in 95 probands with primary lymphedema; onset in elderly male; family history included mother and daughter with lymphedema | [32] |
c.8446C>T | p.Gln2816* | Among 60 patients with primary lymphedema, CELSR1 most frequently mutated in 27 patients. c.8446C>T detected in mother/daughter; classified as “likely pathogenic” | [33] |
c.8871_8872del | p.Cys2957* | Found in daughter; may account for more severe disease course | [33] |
c.4326_4332del | p.Thr1443Glyfs*14 | Non-syndromic lymphedema in a young male: right arm oedema at 9 months, right leg oedema at 19 years. Patient and mother had CELSR1 deletion causing frameshift and premature stop codon. | [34] |
Table 2: Experimental studies describing the genetic features and clinical-phenotypic characteristics of CELSR1-associated lymphedema. Both our patients demonstrate hypoplasia and aplasia of lymphatic vessels in lower extremities. Such a structure of the lymphatic system, onset of lymphedema at birth or in infancy without syndromic features and systemic involvement is associated with the number of conditions, including Lymphatic malformation 1 (Milroy’s lymphedema, OMIM 153100, 5q35.3, FLT4), Lymphatic malformation 2 (OMIM 611944, 6q16.2-q22.1), Lymphatic malformation 3 (OMIM 615907, 1q42.13, GJC2), Lymphatic malformation 4 (OMIM 615907, 4q34.3, VEGFC), Lymphatic malformation 5 (OMIM 153200, not mapped), Lymphatic malformation 9 (OMIM 619319, 22q13.31 CELSR1), Lymphatic malformation 10 (OMIM 619369, 8p23.1, ANGPT2). Aplasia or hypoplasia of initial lymphatic vessels is well-known clinical feature of Milroy’s lymphedema, as well as blood vessel abnormalities, including prominent leg veins and saphenous venous reflux [35–37]. In our study, both siblings have been presented by aplasia of lymphatic vessels according to MRL data (Figure 2). Additionally, male patient had prominent vein on left leg, however nail abnormalities or venous insufficiency were not identified in any of the children. Clinical features of another PLD types are much less studied, so it’s not possible to make a more accurate comparison in phenotypes. However, we did not find any pathogenic changes in coding sequences of genes, associated with primary lymphedema, except of CELSR1, in our patients.
5.Conclusions:
Advancements in genetic testing technologies have revolutionized the diagnosis of changes in various genes associated with lymphedema development. The identification of molecular genetic features of primary lymphedema subtypes has enabled healthcare professionals to provide patients with information about their disease and how to manage potential complications effectively[3,38,39]. At the same time, the use of next-generation sequencing technologies in biomedical and clinical research has led to the discovery of numerous variants of uncertain significance, making the interpretation of results challenging and reducing the number of successful genetic diagnoses. In the case of CELSR1, analyzing the functional and clinical significance of genetic variations is complicated by the fact that the mechanisms of activation and regulation of the receptor remain poorly understood, and the frequency of detectable mutations is very low. Several pathogenic and likely pathogenic variants of CELSR1 have been previously associated with primary lymphedema progression. In our study, for the first time, a substitution in the 7-TM domain of the CELSR1 was revealed in the familiar case of primary lymphedema. To date variant cannot be confidently classified as pathogenic and requires further investigations but might be considered during molecular genetic examination of DNA samples of patients with primary lymphedema.
Author Contributions:
Conceptualization, R.M., Yu.F. and A.R.; methodology, Yu.F. and R.M..; validation, A.R. and R.M.; formal analysis, A.I., E.B., M. Zh.; investigation, Yu.F., A.F., A.I..; resources, A.R.; writing—original draft preparation, Yu.F., A.I.; writing—review and editing, R.M., M.Zh. and A.R; visualization, A.F.; supervision,
R.M. and A.R.; funding acquisition, A.R. All authors have read and agreed to the published version of the manuscript.
Funding:
The work was carried out at the expense of a subsidy allocated to the Kazan Federal University to fulfill the state task in the field of scientific activity (project № FZSM-2023-0011).
Institutional Review Board Statement: The study was conducted in accordance with the Declaration of Helsinki, and approved by the local ethical committee of the Kazan Federal University (2020-12-28, #27).”
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement:
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Acknowledgments:
This work was done within the framework of the Strategic Academic Leadership Program (PRIORITY-2030) of Kazan Federal University.
Conflicts Of Interest:
The authors declare no conflicts of interest.
References
- Arnold M, Numanoglu A: Caustic ingestion in children—a review. Insemped surg. 2017 Apr 1 (Vol. 26, No. 2, pp. 95-104). WB Saunders.
View at Publisher | View at Google Scholar - Wesseling C, Keifer M, Ahlbom A, et al: Long-term neurobehavioral effects of mild poisonings with organOPPhosphate and n-methyl carbamate pesticides among banana workers. Int J Occup Environ Health. 2002 Jan 1;8(1):27-34.
View at Publisher | View at Google Scholar - Khayal EE, Abd El Rady I, Morsi A. Trends and Acute Poisoning Profiles: 2023 Annual Report of Zagazig University Poisoning Treatment Unit. Egyptian Society of Clinical Toxicology Journal. 2024 Jun 1;12(1):45- 59.
View at Publisher | View at Google Scholar - Sánchez-Santed F, Colomina MT, Hernández EH: OrganOPPhosphate pesticide exposure and neurodegeneration. Cortex. 2016 Jan 1;74:417-26.
View at Publisher | View at Google Scholar - Paulus FW, Ohmann S, Möhler E, et al: Emotional dysregulation in children and adolescents with psychiatric
View at Publisher | View at Google Scholar - Elagamy SE, Gabr HM: Predictors of the need for Intensive Care Unit admission in acute organOPPhosphorus poisoning: One year prospective study. Egypt J Forensic SciApplToxicol. 2019 Dec 1;19(4):1-9.
View at Publisher | View at Google Scholar - Kamha AA, Al Omary IY, Zalabany HA, et al: OrganOPPhosphate poisoning in pregnancy: a case report. Basic Clin Pharmacol. 2005 May;96(5):397-8.
View at Publisher | View at Google Scholar - Daley SF, Avva U. Pediatric Dehydration. [Updated 2024 Jun 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from:
View at Publisher | View at Google Scholar - Arakawa R, Ito H, Takano A. Paliperidone–Selected References. Aging. 2017;34:211-9.
View at Publisher | View at Google Scholar - Meyer S, Eddleston M, Bailey B, et al. Unintentional household poisoning in children. Klinische Pädiatrie. 2007 Sep;219(05):254-70.
View at Publisher | View at Google Scholar - Boedeker W, Watts M, Clausing P, et al. The global distribution of acute unintentional pesticide poisoning: estimations based on a systematic review. BMC public health. 2020 Dec;20:1-9.
View at Publisher | View at Google Scholar - Tefera GM, Teferi LG. Prevalence, predictors and treatment outcome of acute poisoning in Western EthiOPPia. OPPen access emergency medicine. 2020 Nov 12:365-75.
View at Publisher | View at Google Scholar - Carey JL, Dunn C, Gaspari RJ: Central respiratory failure during acute organOPPhosphate poisoning. Respir PhysiolNeurobiol. 2013 Nov 1;189(2):403-10.
View at Publisher | View at Google Scholar - Razwiedani LL, Rautenbach PG: Epidemiology of organOPPhosphate poisoning in the Tshwane District of South Africa. Environ. Health Insights. 2017 Feb 24;11:1178630217694149.
View at Publisher | View at Google Scholar - Bereda G. Overview of acute poisoning. Arch Epid Pub Health. 2021; 3: 1-2.
View at Publisher | View at Google Scholar - Povey AC: Gene–environmental interactions and organOPPhosphate toxicity. Toxicol. 2010 Dec 30;278(3):294-304.
View at Publisher | View at Google Scholar - Jokanović M, Kosanović M: Neurotoxic effects in patients poisoned with organOPPhosphorus pesticides. Environ. Toxicol. Pharmacol. 2010 May 1;29(3):195-201.
View at Publisher | View at Google Scholar - Hulse EJ, Haslam JD, Emmett SR, et al: OrganOPPhosphorus nerve agent poisoning: managing the poisoned patient. Br JAnaesth. 2019 Oct 1;123(4):457-63.
View at Publisher | View at Google Scholar - Yadav B, Kaur S, Yadav A, et al. Implications of organOPPhosphate pesticides on brain cells and their contribution toward progression of Alzheimer's disease. Journal of Biochemical and Molecular Toxicology. 2024 Mar;38(3):e23660.
View at Publisher | View at Google Scholar - Fernando ME, Lazzarini PA. Neurological Disorders in the Lower Extremity. Neale's Disorders of the Foot and Ankle E-Book: Neale's Disorders of the Foot and Ankle E-Book. 2020 Jun 15:115.
View at Publisher | View at Google Scholar - Pereira EF, Aracava Y, DeTolla LJ, et al: Animal models that best reproduce the clinical manifestations of human intoxication with organOPPhosphorus compounds. J PharmacolExpTher. 2014 Aug 1;350(2):313-21.
View at Publisher | View at Google Scholar - Thunga G, Sam KG, Khera K, et al: Evaluation of incidence, clinical characteristics and management in organOPPhosphorus poisoning patients in a tertiary care hospital. J Toxicol Environ Health Sci. 2010 Oct 31;2(5):73-6.
View at Publisher | View at Google Scholar - Bereda G. Poisoning by OrganOPPhosphate Pesticides: A Case Report. Cureus.2022; 14(10): e29842.
View at Publisher | View at Google Scholar
