

Research Article | DOI: https://doi.org/DOI:10.31579/2834-5126/062
Petrochemical Industry Wastewaters Treatment with Sonication and With Some Chemicals and Toxicity Analysis
- Rukiye Öztekin A
- Delia Teresa Sponza A *
Delia Teresa Sponza, Dokuz Eylül University, Engineering Faculty, Department of Environmental Engineering, Tınaztepe Campus, 35160 Buca/Izmir, Turkey.
*Corresponding Author: Delia Teresa Sponza A, Delia Teresa Sponza, Dokuz Eylül University, Engineering Faculty, Department of Environmental Engineering, Tınaztepe Campus, 35160 Buca/Izmir, Turkey.
Citation: Rukiye Öztekin A, Delia Teresa Sponza A, (2024), Petrochemical Industry Wastewaters Treatment with Sonication and With Some Chemicals and Toxicity Analysis, Clinical Trials and Clinical Research, 3(2); DOI:10.31579/2834-5126/062
Copyright: © 2024, Delia Teresa Sponza A. This is an open access article distributed under the creative commons’ attribution license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 01 March 2024 | Accepted: 29 March 2024 | Published: 22 April 2024
Keywords: daphnia magna acute toxicity assay; hydroxyl radicals concentration; microtox acute toxicity test; petrochemical; polycyclic aromatic hydrocarbons (pahs); sonication
Abstract
The petrochemical industry real wastewaters (PCI ww) cannot be easily treated due to its toxic and refractory organic contents. Therefore, this wastewater was treated by sonication alone and with the addition of some additives. The effects of 1 h aeration, increasing nitrogen gas N2(g) sparging (15 and 30 min), pH (4.0, 7.0, 10.0), dissolved oxygen (DO) (2, 4, 6 and 10 mg L-1), H2O2 (100, 500 and 2000 mg L-1), TiO2 (0.1, 0.5, 10 and 20 mg L-1), NaCl (1, 2.5 and 15 g L-1), ferrous iron (Fe+2) (from FeSO4.7H2O) (2, 8 and 20 mg L-1), ferric ions (Fe+3) (from FeCl3.6H2O) (10, 20 and 50 mg L-1), HCO3-1 ions (from NaHCO3) (0.5, 1 and 5 g L-1) and butanol (C4H9OH) (0.1, 0.5 and 2 g L-1) concentrations, was applied to this wastewater at increasing sonication times (60, 120 and 150 min), temperatures (25, 30 and 60oC), sonication frequencies (25, 35, 132 kHz), sonication powers (120, 350, 640 and 3000 W), and sonication intensities (12.73–16.63–22.64–32.60–50.93 and 90.54 W cm-2), respectively. Sonication alone provided 96.90% PAHs, 92.48% CODdis, 94.23% TOC in PCI ww after 150 min, at 60oC, at 640 W, at 35 kHz, at 90.54 W cm-2, respectively. The maximum removal efficiencies for PAHs, CODdis, TOC were 99.68%, 99.68%, 98.35% at NaCl=15 g L-1, at NaCl=15 g L-1 and at DO=10 mg L-1, respectively, in PCI ww. Higher than 90% acute toxicity removals in PCI ww was measured after sonication with Photobacterium phosphoreum and Daphnia magna organisms for the same operational conditions mentioned above. The results of this study showed that sonication alone ana with some additives can be used effectively to treat the toxic and refractory compounds in PCI ww. Microtox acute tocicity test was more sensitive than Daphnia magna acute toxicxity assay.
1. Introduction
In recent years, ultrasound irradiation has received increasing attention for the destruction of organic pollutants in waters and wastewaters [1-3]. The process comprises cyclic formation, growth and subsequent collapse of microbubbles occurring in extremely small intervals of time, and release of large quantities of energy over a small location. Sonochemical degradation in aqueous phase involves several reaction pathways and zones such as pyrolysis inside the bubble and/or at the bubble–liquid interface and hydroxyl radical (OH)-mediated reactions at the bubble–liquid interface and/or in the liquid bulk [3, 4]. The relative importance of the various mechanisms involved primarily depends on the physicochemical properties of the pollutants in question; the process is more selective towards hydrophobic and volatile species that can be degraded easily via pyrolytic reactions, while hydrophilic and less volatile compounds are degraded slowly via hydroxyl radical-induced reactions [1, 2].
Numerous works have demonstrated the efficiency of ultrasounds toward the degradation of a wide variety of organic compounds including estrogens [for instance, 17b-estradiol (b-E2), estrone (E1), 17a-ethynylestradiol (EE2)], endocrine-disrupting chemicals (EDCs) (such as bisphenol A), pharmaceuticals, vegetable oils, nitroorganics (NOCs), trihalomethanes (THMs), aliphatic hydrocarbons, naphthenic hydrocarbons, polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs) organochlorine pesticides (OCPs), sugars, tannins, pectin, lipids, polyphenols and polyalcohols, etc. present in relatively dilute aqueous solution and it appears that the applications of this novel means of reaction in environmental remediation and pollution prevention is unlimited [5-23].
The intensification of the organic matter solubilization induced by the ultrasonic action, can lead to an increase of the bioavailability of some micropollutants to the degrader consortium [24, 25]. Nevertheless, it is of great interest to study such ubiquitous compounds (PAHs, THMs, OCPs, EDCs, NOCs, etc.), because they are widespread in all parts of the environment; air, water, soils and sediments [6, 8, 26-29]
An important refractory family compounds in petrochemical industry wastewater (PCI ww) are the PAHs, which have a hydrophobic character, a low water solubility and less volatile with increasing molecular weight that limit their biodegradation, their hazard potential can be relatively high, thus making their presence in the water cycle both an acute and a chronic risk to human health and environmental quality [30,31]. Most of the time, compounds studied are hydroxylated and/or halogenated substituted aromatics or hydrocarbons, having a water solubility in a mg/l range. Only a few works concern the degradation of compounds such as PAHs [anthracene (ANT), benzo[k]fluoranthene (BkF), pyrene (PY), benzo[a]pyrene (BaP), etc.] in PCI ww for which solubilities are in the ng/ml range [17, 31-37]. These compounds have been listed by the United States Environmental Protection Agency (US. EPA) and the European Unions European Economic Community (EU EEC) since 1979 and 1980, respectively, as priority pollutants [31, 37-41]. Due to associated health concerns, some PAHs are possible or probable human carcinogen such as BaP and all PAHs having four condensed rings [42]. The distribution, fate and destruction of these pollutants are then of special significance, especially in water. PAHs are ubiquitous environmental pollutants with mutagenic properties, which have not been included in the Turkish guidelines for treated waste monitoring programs [43].
As a consequence of their strongly hydrophobic properties and their resistance to biodegradation, PAHs are not always quantitatively removed from wastewaters by activated sludge treatments, which very efficiently relocate them into treated effluents. In recent years, considerable interest has been shown in the application of ultrasound as an advanced oxidation processes (AOPs) for the treatment of hazardous contaminants, such as PAHs in water.34,44,45 The sonication process is capable of effectively degrading PAHs present in dilute solutions, typically in the micro and nano ranges. PAHs were decayed to less toxic and less carcinogenic products and by-products with addition some catalysts during ultrasonic treatment. However, it is notable that none of the studies report the use of ultrasound for the removal of more hydrophobic PAHs compounds typically found in effluents of PCI ww.
In Izmir-Turkey, the petrochemical industry treatment plant wastewaters are treated with conventional activated sludge systems and approximately seventeen hydrophilic, less and more hydrophobic PAHs [naphthalene (NAP), acenaphthylene (ACL), acenaphthene (ACT), fluorene (FLN), phenantrene (PHE), anthracene (ANT), carbazole (CRB), fluoranthene (FL), pyrene (PY), benz[a]anthracene (BaA), chrysene (CHR), benz[b]fluoranthene (BbF), benz[k]fluoranthene (BkF), benz[a]pyrene (BaP), indeno[1,2,3-cd]pyrene (IcdP), dibenz[a,h]anthracene (DahA), benzo[g,h,i]perylene (BghiP)] are released into receiving bodies, since low COD (56%) and PAHs (39–42%) removal efficiencies are observed. Aerobic activated treatment systems are not equipped for PAHs removal efficiently. PAHs residues have been frequently detected in rivers and lakes that receive sewage and industrial effluents supplied by those surface waters [17].
In this study, the effects of sonication and the addition of some chemicals (1 h aeration, N2(g), pH, DO, H2O2, TiO2, NaCl, Fe,+2 Fe,+3 HCO3-1 and C4H9OH) on the removal efficiencies of PAHs, CODdis, TOC in PCI ww were investigated. The toxicity reduction was assayed with Photobacterium phosphoreum and Daphnia magna after sonication and chemical addition processes. Furhermore, the sensitivities of these toxicity organisms were evaluated.
2. Material and Methods
2.1. Sonicator properties
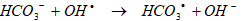
A BANDELIN Electronic RK510 H sonicator was used for sonication of the wastewater samples (Figure 1). The sonication frequency and the sonication power were 35 kHz and 640 W, respectively. Glass serum bottles in a glass reactor were filled to volumes of 100 and 500 ml with wastewater and were placed in a water bath. They were then closed with teflon coated stoppers throughout the measurement of the wastewater. The evaporation losses of PAHs and other volatile compounds were estimated to be 0.01perecentage in the reactor and, therefore, assumed to be negligible. The serum bottles were filled with 0.10 mL methanol in order to prevent adsorption on the walls of the bottles and minimize evaporation.
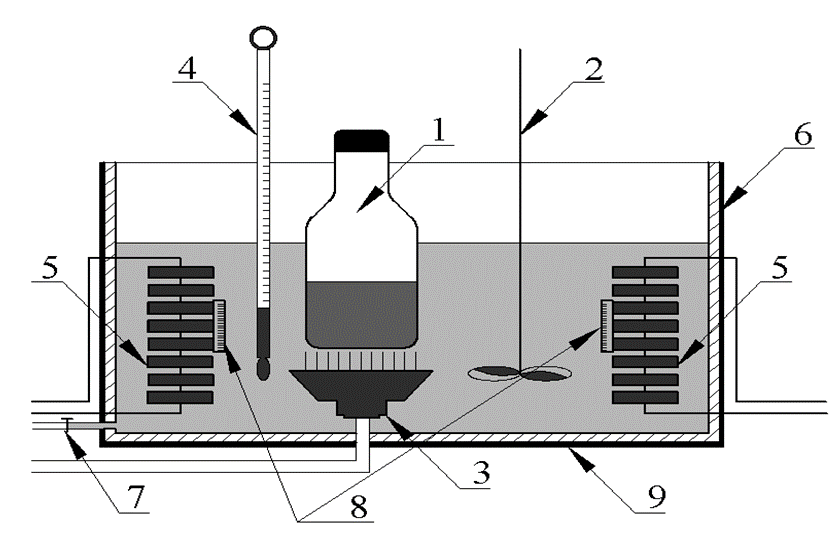
The temperature in the sonicator was monitored continuously and was adjusted to ambient temperature (25oC), 30oC and 60oC with an automatic heater. The stainless steel sonicator was equipped with a teflon holder to prevent temperature losses. The sonicator reactor was sealed with a water
jacket to maintain a constant temperature and to minimize temperature loss. Cooling water was circulated through the jacket of the reactor throughout the experiment in order to remove the heat generated during sonication. This procedure maintains the temperature of the reaction medium almost constant. All experiments were in batch mode using an ultrasonic transducer (horn type), which has an active acoustical vibration area of 19.60 cm2, and a maximum input power of 640 W. Ultrasonic waves for 35 kHz frequency were emitted from the bottom of the reactor through a piezoelectric disc (4.00–cm diameter) fixed on a pyrex plate (5.00–cm diameter). The shematic configuration of the sonicator used in this study is shown in Fig. 1. Samples were taken after 60 min, 120 and 150 min of sonication and they were analyzed immediately.
2.2. Chemical oxygen demand (COD) measurements
COD was determined with Close Reflux Method following the Standard Methods 5220 D 46 using an Aquamate thermo electron corporation UV visible spectrophotometer (2007). First the samples were centrifuged for 10 min at 7000 rpm. Secondly, 2.50 ml volume samples were treated with 1.50 mL 10216 mg L-1K2Cr2O7 with 33.30 g L-1 HgSO4 and 3.50 ml 18.00 M H2SO4 which contains 0.55% (w/w) Ag2SO4. Thirdly the closed sample tubes were stored in a 148°C heater (thermoreactor, CR 4200 WTW, 2008) for 2 h. Finally, after cooling, the samples were measured at 600 nm with an Aquamate thermo electron corporation UV visible spectrophotometer (2007). The Close Reflux Method COD was used to measure the COD in PCI ww before and after sonication experiments. 0.45 μm membrane-filtered (Schleicher and Schuell ME 25, Germany) wastewater samples were used to measure the dissolved COD (CODdis) in PCI ww prior and after sonication experiments.
2.3. Total organic carbon (TOC) measurements
TOC was measured following the Standard Methods 5310 [46] with a Rosemount Dohrmann DC-190 high-temperature total organic carbon (TOC) analyzer (1994).
2.4. Analysis of PAH samples
All extract to analyze for seventeen PAHs with a gas chromotography (Agilent 6890) combined with a mass selective detector (Agilent 5973 inert MSD) (Agilent GC-MS system). A capillary column (HP5-MS, 30.00 m, 0.25 mm, 0.25 μm) was used. The initial oven temperature to hold at 50oC for 1 min, to rise to 200oC at 25oC 1 min-1 and from 200oC to 300oC at 80oC 1 min-1 and was held for 5.50 min. The injector ion source and quadrupole temperatures were 295oC, 300oC and 180oC, respectively. High purity helium (He) was used as the carrier gas at constant flow mode (1.50 mL min-1, 45.00 cm s-1 linear velocity). The MSD to run in selected ion-monitoring mode. Compounds were identified on the basis of their retention times, target and qualifier ions. Qualification was based on the Internal Standard Calibration Procedure.
2.5. Operational conditions
PCI ww samples were examined for different sonication temperetaures (25oC, 30oC and 60oC), different sonication times (30, 60, 120 and 150 min), different sonication frequencies (25–35 and 132 kHz), different sonication powers (120, 350, 640 and 3000 W), different sonication intensities (16.63, 32.60, 50.93 and 90.54 W cm-2), different sonication densities (0.24–0.70–1.28 and 6 W mL-1), respectively. The maximum yields were found 60oC sonication temperature, 150 min sonication time, 35 kHz sonication frequency, 640 W sonication power, 90.54 W cm-2 sonication intensity and 6 W/ml sonication densities, respectively. Therefore, optimum sonication conditions were selected 60oC sonication temperature, 150 min sonication time, 35 kHz sonication frequency, 640 W sonication power, 90.54 W cm-2 sonication intensity and 6 W mL-1 sonication densities, respectively.
Sonication experiments were applied for 1 h aeration, different DO concentrations (2, 4, 6 and 10 mg L-1), different pure N2(g) sparging (15 and 30 min), different pH values (4.0 – 7.0 and 10.0), different H2O2 concentrations (100 mg L-1, 500 and 2000 mg L-1), different TiO2 concentrations ( 0.10 mg L-1, 0.50 mg L-1, 10 and 20 mg L-1), different NaCl concentrations (1 g L-1, 2.5 and 15 g L-1), different Fe+2 ions (from FeSO4.7H2O) concentrations (2 mg L-1, 8 and 20 mg L-1), different Fe+3 ions (from FeCl3.6H2O) concentrations (10 mg L-1, 20 and 50 mg L-1), different HCO3-1 ions (from NaHCO3) concentrations (0.50 g L-1, 1 and 5 g L-1), different C4H9OH concentrations (0.10 g L-1, 0.50 and g L-1), different S2O8-2 concentrations (2 mg L-1, 4 mg L-1, 6 and 10 mg L-1), respectively. Experimental conditions were examined during at 35 kHz, at 640 W, at 60 min, at 120 min, at 150 min, at 30oC, at 60oC ant at 90.54 W cm-2, respectively.
2.6. Microtox acute toxicity assay
Toxicity to the bioluminescent organism Vibrio fischeri was assayed using the Microtox measuring system according to DIN 38412 L34, L341 [47]. Microtox testing was performed according to the standard procedure recommended by the manufacturer [48]. A specific strain of the marine bacterium, Photobacterium phosphoreum Microtox LCK 491 Kit (Dr. LANGE industrial measurement technique in Germany, 2010) were used for Microtox acute toxicity assay. DRLANGE LUMIXmini type luminometer [49] was used for the microtox toxicity assay. Reductions in light intensity at 0., 5th, 15th and 30th min are chosen to measure the toxicity [48, 50]. All samples were serially diluted in 2percentage NaCI (w/v) and each assay was performed at pH=7.0 and a temperature of 15°C. NaCl (2percentage) was used as the control. Samples containing bacterial luminescence were measured for 0 min, 5 min, 15 and 30 min incubation times in a luminometer, respectively. Inhibition percentage (I percentage) values refer to decreasing activity in samples causing inhibitory effect of a test substances and/or a toxic wastewater during the light emission. Color correction was performed according to the DIN 38412 Instructions. The decrease in bioluminescence was indicated the toxic effect of the samples. Toxicity evaluation criteria for luminescent bacteria explained with the percent inhibition effect (H percentage) (Table 1).
| Percent inhibitory effect (H percentage) | Effect |
| 0percentage< H> | Non toxic |
| 5percentage < H> | Moderate toxic |
| 20% < H> | Toxic |
Table 1: Effect of the samples on the inhibition of luminescent bacteria 48.
The decrease of bacterial luminescence due to the addition of toxic substances was calculated as follows in Eqs. (1), (2) and (3):
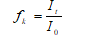
(1) where; fk: Temporary correction factor determined from the control measurements, I0: Initial values of luminescence of control and test sample (0.50 mL bacterial suspension), It: Values measured 0. 5th, 15th and 30th min after 0.5 mL of control or test sample was aded to 0.50 ml of bacterial suspension.
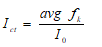
(2) where; Ict: I0 value adjusted by the correction factor, avg fk: The average values of fk.
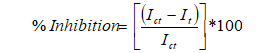
(3) Toxicity evaluation criteria for luminescent bacteria are presented in Table 1. If the percent inhibitory effect (H percentage ) change between 0perecentage and 5percentage, the effect is non-toxic. When H percentage is between 5perecentage and 20perecentage , the effect is possibly toxic, and when H percentage is between 20percentage and 90percentage the effect is toxic [49].
2.7. Daphnia magna acute toxicity test
Toxicity to test using 24 h born Daphnia magna as described in Standard Methods (2005) [46] After preparing the test solution, experiments to carry out using 5 or 10 Daphnids introduced into the test vessel. These vessels to control with 100 mL of effective volume at 7.0-8.0 pH, providing a minimum DO concentration of 6 mg L-1 at an ambient temperature of 20-25oC. Young Daphnia magna to use in the test (in first start ≤ 24 h old). A 24 h exposure is generally accepted for a Daphnia acute toxicity test. Results to express as mortality percentage of the Daphnids. The immobile animals which do not able to move, to determine as dead Daphnids. The results of Daphnia magna acute toxicity test were determined EC (mg L-1) as following Eq. (4):
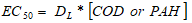
(4) where; EC50: The effective concentration at 50percentage of inhibition ratio (mg L-1or ng mL-1), DL: Inhibition ratio, [COD or PAH]: The COD or PAH concentrations of sonicated sample (mg L-1or ng mL-1).
2.8. Statistical analysis
Differences insensitivity scores between microorganisms were determined by a parametric t-test. The statistical package used for the analysis was SPSSWIN for Windows. The multiple regression analysis between y (dependent) and x (independent) variables was carried out using SPSSWIN for Windows Excel data analysis. The linear correlation was assessed with regression coefficient (R2). The ANOVA (analysis of variance) test was performed in order to determine the statistical significance between x and y variables [49].
Analysis of variance (ANOVA) between experimental data was performed to detect the F and p values. In other words, the ANOVA test was used to test the differences between dependent and independent groups [49]. The comparison between the actual variation of the experimental data averages and standard deviation was expressed in terms of F ratio. F was equal to “found variation of the data averages/expected variation of the data averages.” p reported the significance level. All results are reported at significance levels of p=0.01 and p=0.001. Regression analysis was applied to the experimental data in order to determine the regression coefficient (R2) [50].
Parameters | Removal efficiency (%) | ||
| CODdis (%) | TOC (%) | PAHs (%) | |
| Raw ww, only sonication | 92.48 | 94.23 | 96.90 |
| 1 h aeration | 99.68 | 95.27 (at 30oC) | 97.62 |
| 30 min N2(g) | 96.27 | 96.69 | 96.27 |
| pH = 7.0 | 96.90 | 95.27 (at 30oC) | 96.09 |
| DO = 10 mg L-1 | 97.23 | 98.35 | 97.23 |
| H2O2 = 2000 mg L-1 | 98.04 | 95.88 | 98.04 |
| TiO2 = 20 mg L-1 | 93.88 | 93.20 | 93.88 |
| NaCl = 15 g L-1 | 99.68 | 98.30 | 99.68 |
| Fe+2 = 20 mg L-1 | 98.56 | 95.73 | 98.56 |
| Fe+3 = 50 mg L-1 | 96.76 | 92.45 | 96.76 |
| HCO3-1 = 5 g L-1 | 82.62 | 83.09 | 81.69 (at HCO3-1 = 1 g L-1, 30oC) |
| C4H9OH = 2 g L-1 | 82.62 | 82.75 | 82.62 |
Table 2: The removal efficiencies in PCI ww during sonication process with only sonication and with the addition of some chemicals at 35 kHz, 640 W, at 60oC after 150 min sonication.
The maximum 93.88% CODdis, 93.20% TOC, 93.88% total PAHs yields in PCI ww were observed with TiO2=20 mg L-1 at 60oC after 150 min. The maximum 99.68% CODdis, 98.30% TOC, 99.68% total PAHs removals in PCI ww were measured with NaCl=15 g L-1at 60oC after 150 min. The maximum 98.56% CODdis, 95.73% TOC, 98.56% total PAHs yields in PCI ww were obtained with Fe+2=20 mg/l at 60oC after 150 min. The maximum 96.76% CODdis, 92.45% TOC, 96.76% total PAHs removals in PCI ww were observed with Fe+3=50 mg L-1 at 60oC after 150 min. The maximum 82.62% CODdis, 83.09% TOC yields in PCI ww were measured with HCO3-1=5 g L-1 at 60oC after 150 min. The maximum 81.69% total PAHs removal in PCI ww were observed with HCO3-1=1 g L-1 at 30oC after 150 min. The maximum 82.62% CODdis, 82.75% TOC, 82.62% total PAHs removals in PCI ww were found with C4H9OH=2 g L-1 at 60oC after 150 min (Table 2).
The maximum CODdis removal yield was 99.68% in PCI ww at 1 h aeration and at NaCl=15 g L-1 and at 60oC after 150 min sonication (Table 2). The maximum TOC removal yield was 98.35% in PCI ww at DO=10 mg L-1 and at 60oC after 150 min sonication. The maximum PAHs removal yield was 99.68% in PCI ww at NaCl=15 mg L-1 and at 60oC after 150 min sonication (Table 2).
3.2. Effect of DO concentrations on the removal of PAHs in PCI ww at increasing sonication times and temperatures.
The raw PCI ww samples were oxygenated with increasing DO concentrations (2 mg L-1, 4 mg L-1, 6 and 10 mg L-1) with pure O2 before the sonication experiments. 90.22%, 92.27%, 93.77% and 94.32% total PAHs removal efficiencies were measured in 2 mg L-1, 4 mg L-1, 6 and 10 mg L-1 DO concentrations, respectively, after 150 min at pH=7.0 and at 30oC (Table 2). Only 4 mg L-1, 6 and 10 mg L-1 DO concentrations increased the total PAHs removal efficiencies from 90.11% to 92.27-94.32% in comparison to the non-oxygenated samples (control, E=90.11% total PAHs at pH=7.0) at 30oC after 150 min. In other words, the effect of increasing DO concentrations on the total PAHs removals was found to be insignificant at 30oC after 150 min. The total PAHs yields also increased significantly (from 45.34% to 64.66% and from 62.40% to 80.34%) after 60 and 120 min, respectively, compared to the control at all DO concentrations for the same temperature.
94.67%, 95.00%, 96.79% and 97.23% total PAHs removal efficiencies were obtained in 2 mg L-1, 4 mg L-1, 6 and 10 mg L-1 DO at pH=7.0 and at 60oC after 150 min. As shown, the total PAHs removal efficiencies increased as the sonication time increased. However, the PAHs yields did not show a significant increase at increasing DO concentrations compared to the control (DO=0 mg L-1 while E=96.90% total PAHs at pH=7.0 and at 60oC). The maximum total PAHs removal efficiency was 97.23?ter 150 min in DO=10 mg L-1 at 60oC. Increasing the temperature also did not increase the total PAHs removal efficiencies at increasing DO (ANOVA, F=2.51, p=0.001).
O2 mediates the rate of mineralization of PAHs. The higher DO content of PCI ww resulted in faster rates of PAHs degradation through sonication, supporting the hypothesis that increased oxygenation was largely responsible for the enhanced PAHs degradation. Although, O2 exposure is an important factor in PAHs degradation it was reported that saturating the solutions cause decreases in O2Hl production resulting in low PAHs yields. In the presence of O2, the reactive radicals (Ol , OHl, OOHl) will be produced by a series of reactions and thus contribute to the removal of the PAHs [31]. Although, O2 and air have similar ratios of specific heats and thermal conductivity, the highest formation rate of H2O2, which was induced from the recombination of reactive radicals (OHl and O2Hl) was observed under O2 [51]. In
oxygenated solutions, the O2Hl formed by Eq. (5);
(5)
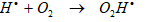
will decay with generation of H2O2. However, the production of the O2Hl increases the oxidation process due to further formation of H2O2 by its recombination reaction Eq. (6) [51]:
(6)
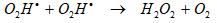
The O2 dissolved in water contributes to the effective formation of many oxidants such as OHl and Ol by pyrolysis of O2. At the same time, these oxidants may further react to produce O2Hl [34, 44]. On the other hand, the maximum temperature produced in collapsing cavitation bubble was higher by sonication in Ar than O2, because of high specific heat ratio of Ar [51] and O2 [52] With the combination of O2 and Ar, it is considered that many oxidants such as O3 and Ol were produced by stronger pyrolytic effect arising from the presence of O2 in addition to OHl typically formed by thermal decomposition of H2O. Moreover, it is also considered that production of OHl was accelerated by the reaction of O2Hl and H2O with oxidants O3 and Ol [53]. Hence, it is assumed that more reactive species in Ar/O2 were formed compared with those in O2/N2 and in Ar. Lawton and Show [54] reported that the decomposition of dichlorinated groups in the Ar/O2 mixture was the highest due to the coupled effects of pyrolysis and radical reaction.
As aforementioned aqueous phase sonolysis is likely to result in the formation of H2O2 which may be formed through the recombination of OHl at the gas–liquid interface and/or in the solution bulk. Moreover, if the solution is saturated with O2, (H2O) and more OHl are formed in the bubble, the recombination of the former at the interface and/or in the solution bulk results in the formation of additional H2O2 [55]. It was found that PAHs mainly decomposed at the gas–liquid interface through both OHl oxidation and pyrolytic reactions (for instance, CH4 and CO2 were identified as the primary pyrolysis by-products) rather than in the solution bulk [56]. This was attributed to the fact that the PAHs, in this study, although less water soluble and non-volatile molecules, tended to accumulate at the gas–liquid interface. Furthermore, it was also found that the concentration of H2O2 formed during PAHs sonication experiments was substantially lower than that formed during water sonication in the absence of PAHs.
3.3. Effect of aeration on the removal of PAHs in PCI ww at increasing sonication time and temperature.
PCI ww were aerated for 1 h with an air pump before the sonication experiments. 48.95%, 75.39% and 94.48% total PAHs yields were measured under 1 h aeration after 60 min, 120 and 150 min, respectively, at pH=7.0 and at 30oC (Table 2). The contribution of aeration were 3.61%, 12.99% and 4.37% to the total PAHs yields after 60 min, 120 and 150 min, respectively, at pH=7.0 and at 30oC, compared to the control (E=45.34%, 62.40% and 90.11% total PAHs after 60 min, 120 and 150 min, respectively, at pH=7.0 and at 30oC).
54.21%, 80.16% and 97.62% total PAHs removals was obtained under 1 h aeration at 60oC after 60 min, 120 and 150 min, respectively, while the control has a yield of 96.90?ter 150 min. Aeration did not increase the total PAHs yields after 60 and 120 min compared to the control at 60oC. The maximum total PAHs yield was 97.62?ter 150 min under 1 h aeration at 60oC. The contribution of aeration to the total PAHs removal was not significant at 60oC (R2=0.45, F=4.56, p=0.01).
The aeration contributed only to the removals of less hydrophobic PAHs. The yields in BbF, BkF and BaP increased from 62.15-67.21% to 93.94-97.99% with 1 h aeration after 150 min at 60oC. Air in the aqueous solution was reported to play a very important role in the generation of highly oxidative OHl enhancing the decomposition of less hydrophobic PAHs [51]. In the presence of air, reactive radicals such as O●, OH● and O2H● will be produced by a series of reactions and may participate in the decomposition reaction of the less hydrophobic PAHs. These PAHs are degraded with hydroxylation reactions since the OHl ions concentrations increased from 23x10-21 up to 8x10-7 ng mL-1after 120 min under 1 h aeration. However, aeration did not contribute to the yields of more hydrophobic PAHs.
In this study, it was found that, under pyrolytic conditions, the H2O2 formation from the reactive radicals (OH● and O2H●) decreased trough sonication of more hydrophobic IcdP, DahA and BghiP. The studies including the H2O2 generation in deionized water showed that H2O2 accumulated in deionized water (pH=2.0) and increased up to 148 mg L-1 with 1 h aeration after 120 min at 30oC (Table 3).
PAHs | H2O2 Production (mg L-1) | OHl Concentration (ng mL-1) |
PAH * | |||||
| Deionized water | ||||||||
| BS | BS | AS-1 | BS | AS-1 | BS | AS-1 | ||
| BbF | 4 | 178 | 5 | 148 | 148 | 23 x 10-21 | 8 x 10-7 | 92 |
| BkF | 4 | 178 | 5 | 148 | 148 | 23 x 10-21 | 8 x 10-7 | 90 |
| BaP | 5 | 178 | 5 | 148 | 148 | 23 x 10-21 | 8 x 10-7 | 89 |
| IcdP | 5 | 178 | 5 | 148 | 148 | 23 x 10-21 | 4 x 10-20 | 47 |
| DahA | 5 | 178 | 5 | 148 | 148 | 23 x 10-21 | 5 x 10-19 | 56 |
| BghiP | 5 | 178 | 5 | 148 | 148 | 23 x 10-21 | 6 x 10-20 | 56 |
| *: PAH removal efficiency; BS: Before sonication; AS-1: After 120 min | ||||||||
Table 3: H2O2 production, OHl ion concentrations through sonication of BbF, BkF, BaP, IcdP, DahA and BghiP before and after 120 min at 30oC (640 W, 35 kHz, initial CODdis =1027.43 mg L-1, initial TOC=620.81 mg L-1, initial total PAHs=1378.77 ng mL-1, n=3, mean values).
The yield of H2O2 decreased to a value as low as 5 mg L-1 from 178 mg L-1 under sonication of less hydrophobic PAHs after 150 min, whereas it could only cause the destruction of some less hydrophobic PAHs (BbF, BkF and BaP) with a yield of 67-92% in comparison with the more hydrophobic PAHs removed (IcdP, DahA and BghiP) with a yield of (47-56%) (Table 3). The PAHs containing high benzene rings were removed with low efficiencies compared to the low benzene ring PAHs (BkF, BaP) since their solubilities, Henry’s law constant (5.84x10-7 and 4.57x10-7 atm m3 mol-1 at 25oC) and vapor pressures (9.70x10-10 and 5.49x10-9 mm Hg at 25oC) are low. The low removal efficiencies in more hydrophobic PAHs (IcdP, DahA) could probably be attributed to their non-hydroxylated sonication mechanism since the OHl ions produced through aeration are not favor for their sono-degradation [17, 57]. A non-hydroxylated pathway was observed in the work of Psillakis et al. [17] with aeration in the removal of some more hydrophobic PAHs (IcdP, DahA) at low frequency. In the presence of more hydrophobic PAHs (IcdP, DahA) more aeration inside the bubble dissolves into the medium during oscillations with a consequent rise in the intensity of collapse and radical production. Higher concentration at the bubble interface raises the partial pressure of the more hydrophobic PAHs (IcdP, DahA) which leads to entrapment of these PAHs into the cavitation bubble, resulting in pyrolytic sono-decomposition during the transient collapse of the bubble [58]. The less hydrophobic (BkF, BaP) PAHs could not be destroyed under aerated sonication process since it can not be removed in hydroxylated mediums containing OHl ions as high as 12x10-8 ng mL-1 [59]. The more hydrophobic BghiP could not be destroyed in hydroxylated mediums containing OHl ions as high as 23x10-21 and 8x10-7 ng mL-1 (Table 3). The destruction way of this PAH was mainly pyrolysis [59].
The effect of air on the removal of short chain, less hydrophobic, PAHs could be explained as follows: the hydroperoxyl radicals (O2H●) formed by Eq. (7) [59];
(7)
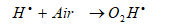
will decay with generation of H2O2 as follows in Eqs. (8), (9) and (10) [59]:
(8)
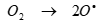
(9)
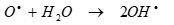
(10)
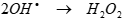
In the presence of air, reactive radicals such as O●, OH● and O2H● will be produced by a series of reactions and may participate in the decomposition reaction of the PAHs. In aerated solutions, the O2H● formed by Eq. (7) given in the section “effect of DO on the sonication of PAHs” and will decay with the generation of H2O2. Air in aqueous solution of less hydrophobic PAHs was reported to play a very important role in the generation of highly oxidative OHl, enhancing its decomposition [59]. The effect of air on the removal of short chain, less hydrophobic, PAHs could be explained as follows: In aerated solutions, O2H● formed by Eq. (8).
The production of the O2H● increases the oxidation process due to further formation of H2O2 by its recombination reaction [51, 57, 60-66]. This was attributed to increase OH entering to the bulk solution. It has been speculated that the rate of OH formation in the gas phase is higher in an oxygenated atmosphere. Since in this study it was studied at low frequency (35 kHz) longer collapse times for the bubbles OHl have more time to recombine before being ejected from inside the bubble into the bulk liquid than at higher frequencies [67]. This implies that OHl reacts with less hydrophobic PAHs primarily in the bulk solution and that the number of OHl capable of reaching the bulk at 35 kHz. It is clear that the destruction yields of less hydrophobic PAHs increases significantly up to 99% with an increase of aeration from 1 h up to 2 h [51]. This observation may be partially ascribed to the generation of OHl, resulted from dissociation of molecular O2 (in the air) in the bubble, which is likely to recombine to form H2O2 at the gas–liquid interface of the bubbles as given in Eqs. (8), (9) and (10).
3.4. Effect of N2(g) on the PAHs removal efficiencies in PCI ww versus sonication times and temperatures.
15 and 30 min N2(g) (3 and 6 mg L-1 N2) was sparged in PCI ww before sonication experiments. Maximum 92.46%, 94.16% and 88.33%, 96.27% total PAHs removal efficiencies were found under 15 and 30 min of N2(g) (3 and 6 mg L-1 N2) sparging at 30oC and 60oC, respectively, after 150 min at pH=7.0 (Table 4). An increase of 6.08-13.18% and 18.51-18.71% in total PAHs yields were obtained under 15 and 30 min N2(g) (3 and 6 mg L-1 N2) sparging after 60 and 120 min while no significant increase in PAHs removals were observed after 150 min at pH=7.0, compared to the control [without N2(g) E=90.11% for total PAHs after 150 min at pH=7.0 and at 30oC]. The contribution of 15 and 30 min N2(g) (3 and 6 mg L-1 N2) sparging on the PAHs removals are significant at low sonication times (60 and 120 min) and temperature (30oC) (R2=0.84, F=13.09, p=0.001). N2(g) sparging did not significantly affect the PAHs removals compared to the control (E=96.90% total PAHs at pH=7.0) at 60oC at all sonication times (R2=0.32, F=1.34, p=0.001).
| Conditions | H2O2 Conc. (mg/l) (60oC) | OHl ion Conc.(mg/l) |
| H2O2 concentration (mg L-1) in deionized water (60oC) | 187 | 0 |
| H2O2 concentration (mg L-1) for less hydrophobic PAHs (60oC) after 30 min | 198 | 10x10-52 |
| H2O2 concentration (mg L-1) for more hydrophobic PAHs (60oC) after 30 min | 4 | 10x10-62 |
| H2O2 concentration (mg L-1) for less hydrophobic PAHs (60oC) after 120 min | 108 | 2x10-22 |
| H2O2 concentration (mg L-1) for more hydrophobic PAHs (60oC) after 120 min | 4 | 10x10-62 |
| H2O2 concentration (mg L-1) for less hydrophobic PAHs (60oC) after 150 min | 9 | 43x10-7 |
| H2O2 concentration (mg L-1) for more hydrophobic PAHs (60oC) after 150 min | 4 | 10x10-62 |
Table 4: Effect of 30 min N2(g) (6 mg L-1 N2) sparging on H2O2 production and OH ion concentrations in PCI ww at 60oC after 30 min, 120 and 150 min (640 W, 35 kHz, initial CODdis=1027.43 mg L-1, initial TOC=620.81 mg L-1, initial total PAHs=1378.77 ng mL-1, n=3, mean values).
Similar to the results of [52] 15 and 30 min N2(g) (3 and 6 mg L-1 N2) sparging did not contribute to the PAHs removals at 60 and 120 min at 60oC
with the exception of 15 and 30 min N2(g) (3 and 6 mg L-1 N2) sparging after 120 min at 30oC8 (Table 4). This could be attributed to the benzene ring-opening reactions of PAHs and other intermediates as well as the degradation of by-products throughout N2(g) sparging at low temperature.
H2O2 which is preferentially formed by the recombination of OHl issue from the sonolysis of H2O can be used as a good indicator of the OHl production. The H2O2 measurement during acoustic cavitation, in absence and in the presence of PCI ww, is a suitable method to estimate the radical production rate for specific sonochemical conditions. The initial rate of H2O2 formation associated to the PAHs treatment by sonication in PCI ww decreases with increasing sonication time at 60oC. In the absence of PCI ww (in deionized water) the H2O2 was accumulated and its concentration was measured as 187 mg L-1, whereas this level was only 9 mg L-1in PCI ww after 150 min in samples containing N2 (Table 4). The OHl ion concentrations also increased from 10x10-62 to 43x10-7 mg L-1 after 150 min in PCI ww containing less hydrophobic FL, BaA and BbF. This showed that hydroxylation is the main mechanism for the removal of less hydrophobic PAHs. In other words, OHl is the major process for complete degradation of less hydrophobic PAHs.
In PCI ww the most sonogenerated OHl reacted with 89-97% less hydrophobic PAHs removals and radical recombination to produce H2O2 [68]. The OHl ion concentrations remained constant around 10x10-62 mg L-1 after 150 min in PCI ww containing more hydrophobic BaP, IcdP, DahA and BghiP. No increase in OHl ion concentrations was observed throughout sonication of more hydrophobic PAHs. This showed that hydroxylation is not the main mechanism for the removal of more hydrophobic PAHs. This indicates that the main process for the destruction of more hydrophobic PAHs is pyrolysis.
In other words, in this study, the contribution of OHl is minor for the ultimate sonodegradation of more hydrophobic PAHs. The formation of by-products (hydroxylated compounds namely phenanthrenediols) for possible OHl oxidation was not observed in HPLC. Similar results were obtained in the studies performed by Lindsey and Tarr [69], Wen et al. [70], Wu and Ondruschka [55]. Since the sonooxidation of less hydrophobic FL, BaA and BbF comprised 0.87% and 0.77% and 79% of the total sonodegradation process, OHl is the major process for complete degradation of these PAHs. The contribution of the pyrolysis to the destruction of these PAHs is not significant.
Different suggestions were reported on the effect of N2(g) sparging on PAHs removals: Dissolved N2 present in aqueous solution might scavenge the free radical attacks to PAHs [56]. Some studies showed that the PAHs are degraded in a N2(g) sparged system with high concentrations of OHl scavenger through sonication [17]. Sparging of N2(g) could change the temperature within the cavitation site or other properties of the cavitation process. Gasses with lower thermal conductivity values such as N2 (18.70 mW mol-1. K-1) increase the temperature inside the cavitation bubble upon collapse because they will allow less heat to the surrounding [17]. The ratio of the specific heat at constant pressure and constant volume (Cp/Cv) plays a role in determining the maximum size of the cavitation bubble [17, 43]. Since the Cp/Cv ratio of N2(g) is high (1.543) the bubble diameter increase during cavitation process resulting in high OHl ions [36, 57]. In this study it was found that N2(g) increase the OHl ions through sonication of less hydrophobic PAHs by increasing the temperature and bubble diameter as reported by Sivasankar and Moholkar [58].
It was found that N2(g) bubbles produce the highest number of OHl in the sonication of less hydrophobic PAHs. Since the intensity of collapse of N2(g) bubbles indicated by the temperature peaks attained at the collapse of these bubbles as reported by Sivasankar and Moholkar [58]. Although, some researchers mentioned that the trend of N2(g) in production of OHl is low through hydroxylation reaction in a medium containing more hydrophobic PAHs the dissolved N2(g) slowly diffuses into the cavitation bubble during oscillations, as result of which the equilibrium of the bubble grows [17, 57]. This gas cushions the transient collapse of the bubble, the temperature and pressure peaks attained in the bubble reduce, resulting in the reduction of OHl production. This affects negatively the degradation of PAHs occurring through hydroxylation. In the presence of more hydrophobic PAHs more N2(g) inside the bubble dissolves into the medium during oscillations with consequent rise in the intensity of collapse and radical production. Higher concentration at the bubble interface raises the partial pressure of the pollutant that leads to higher evaporation and subsequent entrapment of the pollutant into the cavitation bubble, which under goes pyrolytic decomposition at the extreme conditions reached during the transient collapse of the bubble [71]. Sivasankar and Moholkar [58] mentioned that the presence of N2(g) has a negative effect on the formation of H2O2 and on the degradation of PAHs during sonication under air. The HNO2 formed may be the scavenger of OH, which eventually leads to the suppression in H2O2 production.
3.5. Effect of H2O2 concentrations on the removal of PAHs in PCI ww at increasing sonication times and temperatures
100 mg L-1, 500 and 2000 mg L-1H2O2 were added in PCI ww before the sonication experiments. 89.63%, 93.28% and 96.46% total PAHs removals were observed in 100 mg L-1, 500 and 2000 mg L-1 H2O2, respectively, at pH=7.0 and at 30oC after 150 min. An increase of 3.17% and 6.35% in total PAHs removals were measured in 500 and 2000 mg L-1 H2O2, respectively, after 150 min at pH=7.0 and at 30oC, compared to the control (without H2O2 while E=90.11% total PAHs at pH=7.0 and at 30oC). Although, a correlation between PAHs removals and H2O2 concentrations is observed, this relationship is not significant at pH=7.0 and at 30oC after 60 and 120 min at 30oC (R2=0.80, F=2.87, p=0.001).
91.33%, 94.19% and 98.04% total PAHs removal efficiencies were obtained in 100 mg L-1, 500 and 2000 mg L-1 H2O2 at 60oC after 150 min at pH=7.0. No significant increase in total PAHs yields was obtained by increasing the H2O2 concentrations compared to the control (E=96.90% for total PAHs at pH=7.0) at 60oC (R2=0.40, F=3.87, p=0.001). The PAH yields in the samples containing H2O2 and non-containing (control) samples were around 52.61–57.90% and 77.22-82.19%, respectively, at 60oC after 60 and 120 min at pH=7.0. The maximum total PAHs removal efficiency was 98.04?ter 150 min in H2O2=2000 mg L-1 at pH=7.0 and at 60oC.
It was reported that this oxidant elevates the extent of PAHs removal through acustic cavitation [72]. The similar degradation degree in the control and in the presence of H2O2 at 60oC after 120 and 150 min may be attributed to the increased level of OH scavenging by the PAHs and by H2O2 itself. During the sonolysis of aqueous solutions, OH and H are generated by the thermolysis of water in the solution medium and can scavenge OH produced. As the concentration of H2O2 in the solution is increased, it’s OHl scavenging effect increases causing decrease in degradation of PAHs. It was reported that at very high H2O2 concentrations detrimental effects are observed, since the recombination reaction of OHl is more predominant and H2O2 acts as a scavenger for OHl [72]. The scavenging of free OH becomes the dominant process at high H2O2 concentrations in the system, thereby lowering the ability of OHl to degrade PAHs. A similar trend has been reported in the sonolytic destruction of 5,5–dimethyl–1–pyrroline–N–oxide and oxalic acid using H2O2 [73]. The removal yields of all seventeen PAHs were above 95%.
H2O2 is an oxidizing agent. It was reported that this oxidant increases the extent of PAH removal through acustic cavitation [72]. During the sonolysis of H2O, OHl is produced and recombined into H2O2, if no organic compounds such as radical scavengers are present in the H2O. The formed H2O2 and its sonolytic decomposition products would have an effect on the degradation of organic compounds like PAHs during sonication. The H2O2 may be mainly present not inside the cavitation bubbles but in the bulk solution due to the high H2O solubility and low volatility [74]. In our study, no significant total PAHs yields were obtained by the addition of H2O2. The scavenging of free OHl becomes the dominant process at high H2O2 concentrations in the system, thereby lowering the ability of OH to degrade PAHs [72]. It is also possible that the presence of high H2O2 concentrations results in a lower intensity of cavitation, due to the fact that vaporous cavities will be generated in this case, as reported by Rae et al. [73]. This is not valid for our study since it was perfomed at high sonication intensity.
Since less hydrophobic PAHs are considerably non-volatile with low Henry’s law constant and solubility, [75] sono-degradation inside the cavitation bubble is expected to be insignificant. Therefore, hydroxyl radical-induced reactions are likely to be the main degradation mechanism of less hydrophobic ones as follows in Eqs. (11) and (12):
(11)
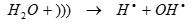
(12)
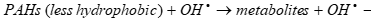
It should be pointed out that OHl, formed via water sonolysis, can partly recombine yielding H2O2 which in turn reacts with H2 to regenerate OHl in Eqs. (13) and (14):
(13)
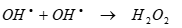
(14)
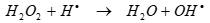
The formation of H2O2 during ultrasound irradiation was confirmed experimentally during sonication in the absence and presence of less and more hydrophobic PAHs. As the H2O2 concentration increased to 176 mg L-1 after 60 min the less hydrophobic PAHs removals increased to 96-98% while the removals of more hydrophobic PAHs remained around 60.24-62.32?ter 120 min (Table 5). This process ends with decreasing of H2O2 to 5 mg L-1 As BbF, BkF, BaP and its degradation metabolites scavenge OHl (Eqs. 13 and 14), the yield, defined as H2O2 formed in the presence of less hydrophobic PAHs over H2O2 formed in deionized water.
| H2O2 Concentration (mg L-1) | Less Hydrophobic PAHs | More Hydrophobic PAHs | ||||||
| in deionized water | After 60 min sonic. | After 120 min sonic. | BbF | BkF | BaP | IcdP | DahA | BghiP |
| 13 | 156 | 5 | ||||||
| Removal eff (%) after 30 min | 34.21 | 32.44 | 38.10 | 19.40 | 21.12 | 20.28 | ||
| Removal eff (%) after 120 min | 95.35 | 96.76 | 97.65 | 76.33 | 75.35 | 77.32 | ||
Table 5: Effect of H2O2 production on the removals of less (BbF, BkF, BaP) and more (IcdP, DahA, BghiP) hydrophobic PAHs after 60 and 120 min at 30oC (640 W, 35 kHz, initial CODdis=1027.43 mg L-1, initial TOC=620.81 mg L-1, initial total PAHs=1378.77 ng mL-1, n=3, mean values).
3.6. Effect of TiO2 concentrations on the PAHs removal efficiencies in PCI ww at increasing sonication times and temperatures.
In order to optimize the addition of an amount of TiO2 catalyst for the highest sonocatalytic destruction of PAHs 0.1 mg L-1, 0.5 mg L-1, 10 and 20 mg L-1 TiO2 was added to the PCI ww before the sonication experiments. In general, the results of this study showed that as the sonication time increased the total PAHs yields also increased at pH=7.0 and at 30oC. All the TiO2 concentrations increased the total PAHs removals after 60 and 120 min, respectively, compared to the control reactor containing no TiO2. The increase of TiO2 concentrations from 0.1 mg L-1 up to 20 mg L-1increased the total PAHs removal after 60 and 120 min at pH=7.0 and at 30oC. It was found that the total PAHs yields were the same as the control reactor free of TiO2 and the reactor containing 20 mg L-1 TiO2 after 150 min at pH=7.0 and at 30 oC. The total PAHs removals were slightly lower in the reactors containing 0.1 and 0.5 mg L-1 TiO2 concentrations after 150 min, compared to the control.
As the sonication time was increased from 60 min to 150 min the total PAHs removal increased in all reactors containing TiO2 and in the control reactor free of TiO2 at pH=7.0 and at a temperature of 60oC. No significant increase in total PAHs yield was obtained by increasing the TiO2 concentrations from 0.1 mg L-1 up to 20 mg L-1 at 60oC. The maximum total PAHs removal efficiency was 93.88?ter 150 min in TiO2=20 mg L-1 at pH=7.0 and at 60oC. The increase of TiO2 catalyst did not further decompose the PAHs in aqueous solution. Excessive TiO2 particles sometimes result in mutual screens among TiO2 particles, which not only protect the PAHs molecules, but also reduce the sonocatalytic activity of TiO2 powder, at 32 kHz and at 480 W after 150 min at 60oC as reported by Wen et al. [70]. In this study it was found that high temperature decreased the sonodegradation of total PAHs assisted by TiO2, as reported by Wang et al. [76]. Both sonocatalytic and ultrasonic destructions in the presence of TiO2 decreased gradually along with the increased temperatures [77].
In general, for most of the chemical reactions, the higher the temperature in the reaction system is, the quicker the reaction rate becomes and although, it is known that radical reactions do not depend very much on the systemic temperature. However, the acoustic cavitation which produces the holes on the surface of TiO2 particles or OH in aqueous solution depends on the change of systemic temperature. It is well known that both sonocatalytic and ultrasonic degradation of organic pollutants are related to the acoustic cavitation. The acoustic cavitation can give rise to holes with strong oxidability on the surface of TiO2 particles which either can directly decompose the organic pollutants adsorbed on the surface of TiO2 particles or indirectly remove the organic pollutants in the aqueous solution through the OH resulting from hole oxidation of H2O molecules. When the temperature in the aqueous solution becomes high, the vapor or gas bubbles escape rapidly from the reaction system and thus do not grow or collapse, which badly weakens the acoustic cavitation.
The results of this study showed that the PAHs removals were not dependent on the physicochemical properties of the PAHs mentioned above during sonication enhanced by 20 mg/l TiO2 (R2=0.45, P=4.67, p=0.001). High PAHs removals were also valid for the individual PAHs concentrations measured in a control containing no TiO2 after 150 min. It can be concluded that if PCI ww containing PAHs is sonicated for 150 min at 60oC, it could be removed efficiently without TiO2. As a result, the obtained removal for seventeen PAHs was nearly the same with 20 mg L-1 TiO2 and without TiO2 at 60oC after 150 min.
3.7. Effect of NaCl concentrations on the PAHs removal efficiencies in PCI ww at increasing sonication times and temperatures
Increasing NaCl concentrations (1 g L-1, 2.5 and 15 g L-1) were added in PCI ww before sonication experiments. At 30oC, 90.13%, 92.14% and 99.23% of total PAHs removals were observed in 1 g L-1, 2.5 and 15 g L-1 NaCl after 150 min at pH=7.0 and at 30oC. The total PAHs yields were increased by 2.03% and 9.12% in 2.5 and 15 g L-1 NaCl, respectively, after 150 min at pH=7.0 and at 30oC, compared to the control (without NaCl while E=90.11% total PAHs at pH=7.0 and at 30oC). A significant linear correlation between total PAHs yields and increasing NaCl concentration was not observed (R2=0.38, F=0.31, p=0.01).
93.17%, 96.08% and 99.68% of total PAHs removals were obtained in 1 g L-1, 2.5 and 15 g L-1 NaCl, respectively, after 150 min at pH=7.0 and at 60oC. The contribution of NaCl to the PAHs removal was 2.78% in 15 g L-1 NaCl after 150 min at pH=7.0 and at 60oC, compared to the control (E=96.90% total PAHs at pH=7.0 and at 60oC). The maximum total PAHs removal efficiency was 99.68?ter 150 min in NaCl=15 g L-1 at pH=7.0 and at 60oC. A significant linear correlation between total PAHs yields and increasing NaCl concentration was not observed (R2=0.33, F=0.215, p=0.01).
The contributions of the last two NaCl concentrations to the PAHs removals were only 2.80% and 3.20%, respectively. At NaCl concentrations > 12 g L-1 the partitioning of the PAHs molecules between interfacial and bulk region increases. Besides, addition of salt in the liquid medium reduces the DO concentration and hence, the scavenging of the radicals [78]. As a result, the probability of interaction between PAHs molecules and radicals decrease. The overall effect is reduction in the extent of degradation.
High NaCl concentrations (10-12 g L-1) increase the ionic strength of the aqueous phase, driving the PAHs to the bulk–bubble interface through sonication [79]. This increases the partitioning of the PAHs species upon cavitation implosion in a sonicator resulting in increased surface tension of the PAHs. The higher NaCl concentrations (10-12 g L-1) resulting in high removals can be explained by the fact that a higher amount of NaCl will create more salting out effect than the lower amount and thus increase the interfacial concentration of the PAHs. All of these factors help to collapse the bubbles more violently, causing high PAHs degradation. Since the Na+1 and Cl-1 concentrations in the sonicator were measured as 1.1 and 1.9 mg L-1, respectively, after sonication, it can be concluded that a large part of 12 g L-1 NaCl reach an non-equilibrium adsorption level at the bubble/solution interface under the sonication conditions used. On the basis of this conclusion, it can be mentioned that the nonequilibrium surface excess values for solutes do not fully equilibrate with the bubble/solution interface during sonication. For the case of hydrophobic compounds, in the presence of excess NaCl, an acoustic bubble in a multi bubble field has a finite lifetime, and that this lifetime increases with decreasing applied frequency. This resulting in a adsorption of excess salt to the bubble/solution interface during sonication as reported by Sunartio et al. [80]. Therefore, the NaCl remaining from the sonication did not cause serious pollution.
Depending on the nature of contaminants, addition of salt (NaCl) to the solution can decrease their solubility and consequently increase their hydrophobicity due to the salting-out effect. This is expected to enhance diffusion of solutes from the bulk solution to the bubble–liquid interface, thus leading to increased degradation rates. A possible explanation would be that adding salt to the reaction mixture results in reduced vapor pressure and increased surface tension, both of which tend to reduce the number of bubbles formed [17].
3.8. Effect of Fe+2 concentrations on the removal of PAHs in PCI ww at increasing sonication times and temperatures
2 mg L-1, 8 and 20 mg L-1 Fe+2 ions (from FeCl2.4H2O) were added to the PCI ww before the sonication experiments. 83.27%, 92.62% and 95.12% total PAHs removals were obtained in 2 mg L-1, 8 and 20 mg L-1 Fe+2, respectively, after 150 min at pH=7.0 and at 30oC. No significant increase in PAHs yields was obtained from 8 to 20 mg L-1 Fe+2 after 150 min at pH=7.0 and at 30oC, compared to the control (without Fe+2 while E=90.11% for total PAHs at pH=7.0 and at 30oC). A significant linear correlation between total PAHs yields and increasing sonication time was not observed (R2=0.30, F=0.28, p=0.01).
86.28%, 95.27% and 98.56% total PAHs yields were observed in 2 mg L-1, 8 and 20 mg L-1 Fe+2, respectively, after 150 min at pH=7.0 and at 60oC. No significant increase in total PAHs yields were obtained by increasing the Fe+2 concentrations compared to the control after 120 and 150 min at pH=7.0 and at 60oC. Sonication alone provided 96.90% total PAHs yield after 150 min at pH=7.0 and at 60oC The maximum total PAHs removal efficiency was 98.56?ter 150 min in Fe+2=20 mg L-1 at pH=7.0 and at 60oC. A significant linear correlation between total PAHs yields and increasing Fe+2 concentrations was not observed (R2=0.32, F=0.31, p=0.01).
These high removals in PAHs with high molecular weights could be attributed to effective sonication at 35 kHz with 20 mg L-1 Fe+2. The findings of the study demonstrate that sonication enhanced with 20 mg L-1 Fe+2 can be used to improve the mass transport of poorly soluble PAHs in PCI ww and alleviate limiting steps of removal of hydrophobic refractory PAHs after 150 min at 60oC. Furthermore, the reaction of Fe+2 with H2O2 was formed OH ion. This phenomenon leading to the destruction of benzene rings of hydrophobic and hydrophilic PAHs. Some of the recent studies showed that there is a highly significant relationship between the average removal percentages and the hydrophobicity of PAHs, indicated as the octanol water partition coefficient is shown [55, 81]. It becomes evident that a larger hydrophobicity resulted in smaller removal of the PAHs. However, in this study, the 4-ring, 5 and 6- ring PAHs, in contrast to the relationship mentioned above, exhibiting higher removals than expected from their log Kow at 60oC after 150 min. No significant correlation was observed between PAHs yields, water solubility, vapor pressure, C number in benzene rings, Henry’s law constant through sonication assisted 20 mg L-1 Fe+2 at 60oC after 150 min (R2=0.43, F=3.56, p=0.001).
As reported by Lindsey and Tarr [69] a possible explanation of the positive effect of Fe+2 on the sonication of PAHs could be the reaction of Fe+2 with H2O2 to form OHl at 34 kHz and at 450 W after 125 min at 60oC. As this reaction proceeds, the concentration of Fe+2 declines, and consequently the rate of H2O2 consumption and OHl formation decline. The loss of Fe+2 is eventually balanced by the formation of Fe+2 through reduction of Fe+3 by reaction with H2O2 or O2Hl, and a steady state Fe+2 concentration is reached. At this point (> 60 s), pseudo first order loss of H2O2 is observed. This explanation is also supported by evidence that the OHl formation rate is significantly higher in the first 60 s. Psillakis et al. [17] studied the sono-removal of 150 μg L-1 total initial concentration of PAHs mixture (NAP, ACT, PHE) in an aqueous solution. 92.20% of NAP, 96.25% of ACT and 89.80% of PHE removal efficiencies were found with Fe+2=14 mg L-1 concentration in a sonicator with at 150 W, at 80 kHz, at 20oC, after 150 min. In our study the removal efficiencies for the aforementioned PAHs were found to be higher (E, NAP=99%, E, ACT=98.25% and E, PHE=98.11%) at 35oC for the same sonication time. Beckett and Hua [82, 83] also found that the addition of 0.02-1 ng L-1 Fe+2 concentrations improved the 1,4–dioxane decomposition rate and mineralization efficiency at a 30 kHz and a power of 590 W after 115 min. During aqueous ultrasonic irradiation, OHl formed during the thermolytic reactions of H2O recombine to form H2O2 that tends to accumulate in the solution and does not usually play an important role in oxidizing organic species. However, the reaction between H2O2 and Fe+2 is known to produce OHl and is commonly referred to as the Fenton process [81].
3.9. Effect of Fe+3 concentrations on the removal of PAHs in PCI ww at increasing sonication times and temperatures
Increasing Fe+3 concentrations (10 mg L-1, 20 and 50 mg L-1) were added to the PCI ww before sonication process. 82.92%, 91.72% and 93.58% total PAHs removals were measured in 10 mg L-1, 20 and 50 mg L-1 Fe+3, respectively, after 150 min at pH=7.0 and at 30oC. An increase of 14.66-22.28% and 16.21-20.55% in total PAHs yields were measured for after 60 and 120 min, compared to the control (without Fe+3) at pH=7.0 and at 30oC. Although, a correlation between PAHs removal efficiencies and Fe+3 concentrations were obtained this relationship was not significant (R2=0.76, F=2.56, p=0.01). Control provided 90.11% total PAHs yield after 150 min at pH=7.0 and at 30oC.
84.61%, 93% and 96.76% total PAHs yields were observed in 10 mg L-1, 20 and 50 mg L-1 Fe+3, respectively, after 150 min at pH=7.0 and at 60oC. The contribution of increasing Fe+3 on the total PAHs removal was only 7.06-15.41% and 1.25-5.04% compared to the control after 60 and 120 min at pH=7.0 and at 60oC. The maximum total PAHs removal efficiency was 96.76?ter 150 min in Fe+3=50 mg L-1 at pH=7.0 and at 60oC. However this contribution of Fe+3 was no significant (R2=0.66, F=9.86, p=0.01). Similarly, increasing the Fe+3 concentrations did not significantly affect the PAHs yields compared to the control after 120 and 150 min at pH=7.0 and at 60oC (R2=0.52-0.56, F=8.34-9.91, p=0.01).
In the presence of Fe+3, the sonolytic degradation of less hydrophobic PAHs was enhanced by the increase in OH● induced from the decomposition of the recombined H2O2 [66]. Under these conditions, it can be expected that Fe–O2H+2 as an intermediate produced from the reaction of Fe+3 with H2O2 and it may be partitioned as Fe+2 and O2Hl by the ultrasonic irradiation in Eqs. (15) and (16) [67]. The regenerated Fe+2 also catalyze the decomposition of H2O2 in Eq. (16). These results suggest that in the presence of Fe+3, the sonolytic degradation of less hydrophobic PAHs was enhanced by the increase in OHl [67] in Eq. (17).
(15)
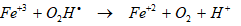
(16)
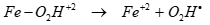
(17)
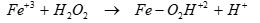
A large fraction of Fe+3 formed from the reaction Fe+2 with H2O2 would mainly exist in the form of Fe+3–O2H or Fe+3–OH complexes. Fe+3 / ultrasound it can be expected that Fe–O2H as an intermediate produced from the reaction of Fe+3 with H2O2 may be partitioned as Fe+2 and O2H● by the ultrasonic irradiation. It was found that the Fe+3/ ultrasound system under O2 is more effective for mineralization than the Fe+2/ ultrasound system and ultrasound only.69 For Fe+3, the overall degradation of organic compounds by oxidation is slower, but the mineralization process is successfully achieved. The degradation of organic compounds by the attack of OH● can be enhanced.
3.10. Effect of HCO3-1 concentrations on the removal of PAHs in PCI ww at increasing sonication times and temperatures
Increasing HCO3-1 (0.5 g L-1, 1 and 5 g L-1) concentrations were added to the PCI ww before sonication process. 80.43%, 92.14% and 81.66% total PAHs yields were found in 0.5 g L-1, 1 and 5 g L-1 HCO3-1, respectively, after 150 min at pH=7.0 and at 30oC. Total PAHs yields were slightly increased in 1 g L-1 HCO3-1 (2%) compared to the control (without HCO3-1 while E=90.11% total PAHs) at pH=7.0 and at 30oC after 150 min. A significant linear correlation between total PAHs yields and increasing sonication time was not observed (R2=0.32, F=0.30, p=0.01).
78.09%, 81.69% and 82.62% total PAHs removals were found in 0.5 g L-1, 1 and 5 g L-1 HCO3-1, after 150 min at pH=7.0 and at 60oC. Total PAHs yields did not change after 150 min compared to the control (E=96.90% total PAHs) at pH=7.0 and at 60oC. A significant linear correlation between total PAHs yields and increasing HCO3-1 concentrations was not observed (R2=0.34, F=0.40, p=0.01).
In order to identify the contribution of OHl in the sonolytic degradation of PAHs, the role of OHl was examined in the absence and presence of HCO3-1. Therefore, one refractory non-volatile, more hydrophobic PAHs namely DahA (4.57 ng/ml) was taken into consideration among the other seventeen PAHs present in PCI ww.
However, significant differences (ANOVA, F=2.67, p=0.001) were observed between the percent degradation values at the acceleration steps. Similar statistical results were obtained between the trials with HCO3-1 and without HCO3-1 after 60 min sonication time (ANOVA, F=1.98, p=0.001 and F=2.80, p=0.001) at the initiation steps. HCO3-1 is a well known OHl scavenger as shown in Eq. (18):
(18)
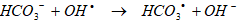
Since HCO3-1 is an ion, it will scavenge free radicals predominantly in the bulk water phase. Water-soluble compounds that are non volatile will be significantly affected by the HCO3-1 concentration [84, 85]. Since DahA is non-volatile, a refractory compound, the HCO3-1 can complete with DahA for available free radicals in the interfacial and bulk region and decrease the sonically destruction efficiency of the PAHs in question. It also shows the contribution of HCO3-1 in the role of OHl scavenger during sonolysis. The effect of radical scavenging by HCO3-1 mainly occurred during the acceleration step, and suggesting that the most OHl in the sonication is generated at the acceleration step. From the results in during the sonication it can be induced that the reaction between OHl and HCO3-1 mainly occurs at the acceleration step rather than at the initial step. As aforementioned OH are mainly produced at the acceleration step, which react with HCO3-1 to produce OHl ion, resulting in the increase in pH of the solution. However, at the initiation step, the production of OHl is insufficient for the reaction with HCO3-1, as shown in Eq. (18). Instead, the presence of 10 mg L-1 HCO3-1 may act as a buffer in the solution, resulting in almost no change of pH.
The sonication of DahA at the initiation step may proceed by a thermal reaction, while the degradation of DahA is dominated by OHl reaction at the acceleration step. Thermal degradation and chemical oxidation contributed approximately 25% and 34% to the degradation of 1,4–dioxane according to the result of the sonication without HCO3-1 as reported by Son et al. [86].
The sonic degradation of DahA in the PCI ww was found to be varied between zero and pseudo first order with respect to PAHs concentration in the initiation and acceleration steps with HCO3-1 and without HCO3-1 at 35 kHz and at 60oC in Eqs. (19) and (20):
(19)
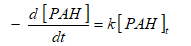
(20)
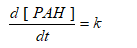
The results of this part of this study showed that the degradation trend of the DahA at the acceleration step was also not significantly different between two conditions with and without HCO3-1 at the beginning of the sonication (0 min). However, the rapid decrease of DahA degradation was observed at the acceleration step in the presence of HCO3-1 after 60 min. The initiation step of the DahA degradation followed the zero order kinetic rate models, while the acceleration step followed the pseudo-first order. In the presence of HCO3-1 as a radical scavenger, the degradations of DahA were suppressed, indicating that OHl is an important factor in the sonication, especially at the acceleration step. Since DahA is hydrophobic, HCO3-1 can compete with DahA for available free radicals in the interfacial and bulk region and decrease its decomposition efficiency. Similar results was also found by Shaw and Lee [87]. HCO3-1 added in the concentration interval 10−3 to 185 mg L-1 acted as OHl scavenger and the expected effect was found to be inhibition of degradation of some dyes and hydrocarbons in pulp and paper industry wastewater. They reported that water soluble compounds that are less volatile will be significantly influenced by the HCO3-1 concentration.
3.11. Effect of iso-butanol (C4H9OH) concentrations on the removal of PAHs in PCI ww at increasing sonication times and temperatures
0.1 g L-1, 0.5 and 2 g L-1 C4H9OH concentrations were added to the PCI ww before sonication process. As the sonication time increased from 60 to 150 min the total PAHs removals increased from 14.41% to 60.14% and 92.14% in 0.1 g L-1, 0.5 and 2 g L-1 C4H9OH, respectively, at pH=7.0 and at 30oC. Sonication alone provided 45.34%, 62.40% and 90.11% total PAHs yields after 60, 120 and 150 min, respectively, at pH=7.0 and at 30oC. In this study, it was found that C4H9OH addition decreased the PAHs yields significantly for 60 and 120 min at pH=7.0 and at 30oC. A significant linear correlation between total PAHs yields and increasing C4H9OH concentrations was not observed (R2=0.32, F=0.43, p=0.01).
No significant contributions of C4H9OH to the total PAHs yields was observed at 60oC. Sonication alone provided 54.21%, 79.31% and 96.90% total PAHs removals after 60, 120 and 150 min, respectively, while the PAHs yields decreased to 15.24%, 60.56% and 82.62% for the sonication given above at 60oC in the samples containing 0.1 g L-1, 0.5 and 2 g L-1 C4H9OH, respectively, at pH=7.0. The maximum total PAHs removal efficiency was 82.62?ter 150 min in C4H9OH=2 g L-1 at pH=7.0 and at 60oC. A significant linear correlation between total PAHs yields and increasing C4H9OH concentrations was not observed (R2=0.34, F=0.38, p=0.01).
Iso-butanol is a known scavenger for the gaseous region and/or interfacial region of the collapsing bubble [88], whereas inorganic salts, such as potassium iodide or bromide reside on the bulk liquid region or possibly at the interfacial region of the cavitation bubble [89]. As the boiling point of iso-butanol (104oC) is only slightly higher than that of water, the addition should not greatly affect the temperature inside the bubble, which is known to depend on vapor pressure [30, 90]. Aqueous sonication occurs in three important regions. The first region is the interior of the collapsing cavitation bubbles; the second region is the interfacial boundary between the gaseous and liquid phases, and the third is the solution bulk [90]. It is possible to obtain information about the sonochemical reaction zones by controlling the type of radical scavenger in which the free radicals are located or produced.
The volatile iso-butanol may scavenge the OHl inside and at the interfacial region of the cavitation bubble and may be partly pyrolyzed inside the cavitation bubble with sufficient ultrasonic irradiation. The cavitation reactions should be influenced by the volatile products formed during the sonication of iso-butanol due to decrease of the temperature inside the collapsing cavitation bubble with long period of irradiation [91-93]. Addition of iso-butanol (740 mg L-1) to the PAHs mixture (FL, BaP, BbF, BkF, BghiP and IcdP) leads to a partial inhibition of the PAHs degradation in agreement with Laughrey et al. [34] and Psillakis et al. [36].
3.12. Effect of pH on the removal of PAHs in PCI ww at increasing sonication times and temperatures
The pH was adjusted to 4.0–7.0 and 10.0 in the PCI ww before sonication process. 91.59%, 90.05% and 92.25% total PAHs removals were found at pH=4.0, pH=7.0 and pH=10.0, respectively, at 30oC after 150 min. Sonication alone provided 90.11% total PAHs removal at 30oC after 150 min at pH=6.98. At short sonication times (60 min and 120 min) the maximum PAHs yields were higher at pH=4.0 and pH=10.0 at 30oC. 63.11%-58.29% and 78.98%-75.70% total PAHs removals was obtained after 60 and 120 min at pH=4.0 and pH=10.0 at 30oC. A significant linear correlation between total PAHs yields and increasing sonication time was not observed (R2=0.31, F=0.33, p=0.01).
93.37%, 96.09% and 95.66% total PAHs removals were observed at pH=4.0, pH=7.0 and pH=10.0, respectively, at 60oC after 150 min. Total PAHs yields did not change after 150 min, compared to the control (at pH=6.98 while E=96.90% total PAHs) at 60oC. The increasing of pH from 4.0 to 10.0 did not significantly affect the total PAHs removals for 60 and 120 min at 60oC. The maximum total PAHs yield was 96.09?ter 150 min at pH=7.0 and at 60oC. A significant linear correlation between total PAHs yields and increasing sonication time was not observed (R2=0.81, F=11.20, p=0.01).
In order to determine the effect of pH on the sonodestruction of PAHs, the pH of the PCI ww was adjusted to acidic (pH=4.0-5.0), neutral (pH=7.0) and alkaline (pH=10.0-11.0) conditions. Figure 2 shows the H2O2 production, OHl ion concentrations and PAHs yields at acidic, neutral and alkaline pH values. The ln OHl ion concentrations decreased in more hydrophobic PAHs (IcdP, DahA, BghiP) (ln -8, ln -23) while they increased in less hydrophobic PAHs (BbF, BkF, BaP) (ln -75, ln -100) at pH=7.0. The H2O2 concentration was high (35-48 mg L-1) through sonication of less hydrophobic PAHs while is low (2-10 mg L-1) through sonodegradation of more hydrophobic PAHs. The high H2O2 production through sonication of less hydrophobic PAHs verified the presence of high OHl ion concentrations as illustrated in Figure 2.
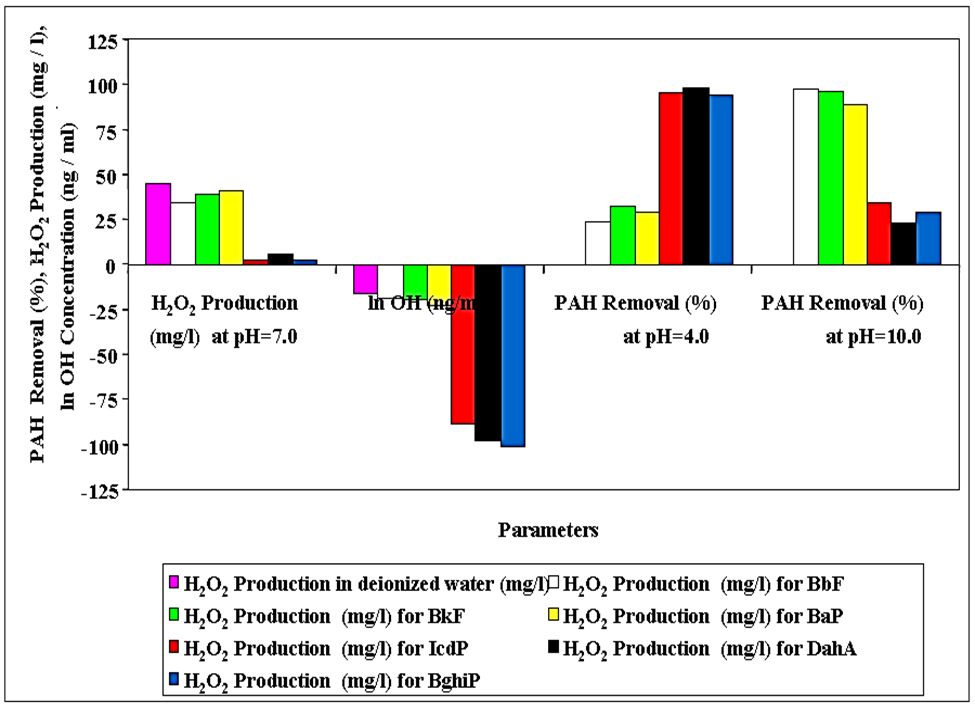
Figure 2: Effects of pH on the removals of PAHs, H2O2 production and OHl ion concentrations (640 W, 35 kHz, initial CODdis=1027.43
| H2O2 Concentration (mg L-1) | Less Hydrophobic PAHs | More Hydrophobic PAHs | ||||||
| in deionized water | After 60 min sonic. | After 120 min sonic. | BbF | BkF | BaP | IcdP | DahA | BghiP |
| 13 | 156 | 5 | ||||||
| Removal eff (%) after 30 min | 34.21 | 32.44 | 38.10 | 19.40 | 21.12 | 20.28 | ||
| Removal eff (%) after 120 min | 95.35 | 96.76 | 97.65 | 76.33 | 75.35 | 77.32 | ||
Table 6: Effect of H2O2 production on the removals of less (BbF, BkF, BaP) and more (IcdP, DahA, BghiP) hydrophobic PAHs after 60 and 120 min at 30oC (640 W, 35 kHz, initial CODdis=1027.43 mg L-1, initial TOC=620.81 mg L-1, initial total PAHs=1378.77 ng mL-1, n=3, mean values).
The formation of H2O2 during ultrasound irradiation was confirmed experimentally during sonication in the absence and presence of less and more hydrophobic PAHs. As the 13 mg L-1 H2O2 increased to 156 mg L-1 after 60 min the less hydrophobic PAHs removals increased to 95.35-97.65% while the removals of more hydrophobic PAHs remained aroud 75.35-77.32?ter 120 min (Table 6). This process ends with decreasing of H2O2 concentration to 5 mg L-1 after 120 min. As BbF, BkF, BaP and its degradation metabolites scavenge OHl (Eqs. 13 and 14), the yield, defined as H2O2 formed in the presence of less hydrophobic PAHs over H2O2 formed in deionized water.
According to work performed by Goskonda et al. [59] the removal of some hydrophobic PAHs from an oil ww by toluene solvent would be significantly enhanced with a reduction of pH values. These PAHs are sonodegraded via pyrolytic reaction that occurs in the hot cavitation bubbles and/or at the gas–liquid interface of the bubbles. It was reported that the concenration of stronger hydrophobic PAHs increased at the gas–liquid interface of the bubbles with an increase of acidity of wastewater. Similarly, Chakinala et al. [72] reported that the sonochemical destruction of hydrophobic compounds like PAHs was enhanced by the addition of salicylic acid with pH < 7>
3.13. PCI ww toxicities, interspecies correlation and sensitivities
The maximum Microtox acute toxicity removal yield was 66.67% in PCI ww after 150 min sonication, at 60oC only with sonication (Table 7). The maximum Microtox acute toxicity removal was 83.33% in PCI ww at 1 h aeration and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 83.33% in PCI ww at 30 min N2(g) (6 mg L-1 N2) sparging and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 66.67% in PCI ww at pH=7.0 and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 77.78% in PCI ww at DO=10 mg L-1 and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 94.44% in PCI ww at H2O2=500 mg L-1 and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 94.44% in PCI ww at TiO2=10 mg L-1 and at 60oC after 150 min sonication (Table 7). The maximum Microtox acute toxicity removal yield was 94.44% in PCI ww at NaCl=2.5 g L-1 and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 88.89% in PCI ww at Fe+2=8 mg L-1 and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 88.89% in PCI ww at Fe+3=20 mg L-1 and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 83.33% in PCI ww at HCO3-1=1 g L-1 and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 88.89% in PCI ww at C4H9OH=0.5 g L-1 and at 60oC after 150 min sonication (Table 7). The maximum Microtox acute toxicity removal yield was 94.44% in PCI ww at H2O2=500 mg L-1, at TiO2=10 mg L-1, at NaCl=2.5 g L-1 and at 60oC after 150 min sonication, respectively (Table 7).
| Removal efficiencies (%) | |||||
| Microtox acute toxicity (%) | Daphnia magna acute toxicity (%) | ||||
| Parameters | 30oC | 60oC | Parameters | 30oC | 60oC |
| 150.min | 150.min | 150.min | 150.min | ||
| Raw ww, control | 55.56 | 66.67 | Raw ww, control | 40.00 | 50.00 |
| 1 h aeration | 77.78 | 83.33 | 1 h aeration | 80.00 | 90.00 |
| 30 min N2(g) | 72.22 | 83.33 | 30 min N2(g) | 70.00 | 90.00 |
| pH=7.0 | 55.56 | 66.67 | pH=7.0 | 40.00 | 50.00 |
| DO=10 mg L-1 | 77.78 | 77.78 | DO=10 mg/l | 70.00 | 90.00 |
| H2O2=500 mg L-1 | 88.89 | 94.44 | H2O2=500 mg/l | 80.00 | 90.00 |
| TiO2=10 mg L-1 | 88.89 | 94.44 | TiO2=10 mg/l | 80.00 | 90.00 |
| NaCl=2.5 g L-1 | 88.89 | 94.44 | NaCl=2.5 g/l | 80.00 | 90.00 |
| Fe+2=8 mg L-1 | 83.33 | 88.89 | Fe+2=8 mg/l | 70.00 | 80.00 |
| Fe+3=20 mg L-1 | 83.33 | 88.89 | Fe+3=20 mg/l | 70.00 | 80.00 |
| HCO3-1=1 g L-1 | 77.78 | 83.33 | HCO3-1=1 g/l | 60.00 | 80.00 |
| C4H9OH=0.5 g L-1 | 77.78 | 88.89 | C4H9OH=0.5 g/l | 60.00 | 80.00 |
Table 7: Acute toxicity removal efficiencies in PCI ww after 150 min sonication at 30oC and at 60oC with the addition of some chemicals for Microtox and Daphnia magna acute toxicity tests (640 W, 35 kHz, n=3, mean values).
The maximum Daphnia magna acute toxicity removal yield was 50% in PCI ww after 150 min sonication, at 60oC only with sonication (Table 7). The maximum Daphnia magna acute toxicity removal yield was 90% in PCI ww
at 1 h aeration and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 90% in PCI ww at 30 min N2(g) (6 mg L-1 N2) sparging and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 50% in PCI ww at pH=7.0 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 90% in PCI ww at DO=10 mg L-1 and at 60oC after 150 min sonication (Table 7). The maximum Daphnia magna acute toxicity removal yield was 90% in PCI ww at H2O2=500 mg L-1 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 90% in PCI ww at TiO2=10 mg L-1 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 90% in PCI ww at NaCl=2.5 g L-1 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 80% in PCI ww at Fe+2=8 mg L-1 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 80% in PCI ww at Fe+3=20 mg L-1 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 80% in PCI ww at HCO3-1=1 g L-1 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 80% in PCI ww at C4H9OH=0.5 g L-1 and at 60oC after 150 min sonication (Table 7).
The maximum Daphnia magna acute toxicity removal yield was 90% in PCI ww at 1 h aeration, at 30 min N2(g) sparging, at DO=10 mg L-1, at H2O2=500 mg L-1, at TiO2=10 mg L-1, at NaCl=2.5 g L-1 and at 60oC after 150 min sonication, respectively (Table 7).
In order to determine the sensitivity of Daphnia magna (water flea) for the acute toxicity tests described and to show the responses of two different organisms, to sonicated and non-sonicated wastewater samples the acute toxicity test was also performed with Vibrio fischeri bacteria (Table 8). Sensitivity ranking indicates the sum of toxicity responses of every organism used in acute toxicity tests [95-101]. To explain the sensitivity of acute toxicity test results based on Daphnia magna and Photobacterium phosphoreum a table was constructed ranking the samples in order of acute toxicity [95-101]. A score of “1” was assigned to the most sensitive test for each sample down to 2 for the least sensitive organism representing two trophic levels which were classified according to the test results [95-101]. The comparison of toxicity response and sensitivity ranking was assessed in PCI ww before sonication process (Table 8).
Parameters | Photobacterium phosphoreum | Daphnia magna | ||
EC50 value and 95% Confidence Limits (mg L-1) 48 h | Sensitivity Ranking | EC50 value and 95% Confidence Limits (mg L-1) 48 h | Sensitivity Ranking | |
Raw ww, only sonication | 16.67 (16.43 – 16.91) | 1 | 25 (24.71 – 25.29) | 2 |
1 h aeration | 4.33 (4.21 – 4.45) | 1 | 5 (4.87 – 5.13) | 2 |
30 min N2(g) | 4.33 (4.21 – 4.45) | 1 | 5 (4.87 – 5.13) | 2 |
pH=7.0 | 16.67 (16.43 – 16.91) | 1 | 25 (24.71 – 25.29) | 2 |
DO=10 mg L-1 | 4.11 (3.99 – 4.23) | 1 | 5 (4.87 – 5.13) | 2 |
H2O2=500 mg L-1 | 2.78 (2.67 – 2.89) | 1 | 5 (4.87 – 5.13) | 2 |
TiO2=10 mg L-1 | 2.78 (2.67 – 2.89) | 1 | 5 (4.87 – 5.13) | 2 |
NaCl=2.5 g L-1 | 2.78 (2.67 – 2.89) | 1 | 5 (4.87 – 5.13) | 2 |
Fe+2=8 mg L-1 | 5.56 (5.42 – 5.70) | 1 | 10 (9.82 – 10.18) | 2 |
Fe+3=20 mg L-1 | 5.56 (5.42 – 5.70) | 1 | 10 (9.82 – 10.18) | 2 |
HCO3-1=1 g L-1 | 8.33 (8.16 – 8.50) | 1 | 10 (9.82 – 10.18) | 2 |
C4H9OH=0.5 mg L-1 | 5.56 (5.42 – 5.70) | 1 | 10 (9.82 – 10.18) | 2 |
Table 8: Sensitivity ranking and EC50 values of PCI ww in Photobacterium phosphoreum and Daphnia magna in sonicated samples containing some additives after 150 min at 60oC (640 W, 35 kHz, n=3, mean values).
The acute toxicity classification of an effluent should always be based on the results of testing all trophic levels at least once. The EC50 value of PCI ww increased from EC50=333.33 mg L-1 (converted from EC90=600 mg L-1 in PCI ww for Microtox acute toxicity assay before sonication) to EC50=618 mg L-1 as the trophic level increased from bacteria in Microtox test to water flea in Daphnia magna test in the influent samples (Table 9). The mean sensitivity scores were 7 and 12 in Vibrio fischeri and Daphnia magna, respectively, after 150 min at 30oC during triplicate sampling for six PAHs. The EC50 values measured for Daphnia magna differed significantly from Photobacterium phosphoreum in the decline of sensitivity scores and exhibited lower mortalities in the influents (t test statistic 3.72, p ≤ 0.05). The t statistics showed that Daphnia magna had higher EC50 values and lower sensitivity scores than Photobacterium phosphoreum. The Photobacterium phosphoreum and Daphnia magna had different responses to toxicity and mortalities (t-test statistic=3.88, p=0.10) and this difference was significant (t test statistic=11.99, p=0.05). The Photobacterium phosphoreum bacteria
had lower EC50 values than Daphnia magna and these differences were significant (t-test statistics=14.42, p < 0 statistic=11.50,> Parameters Microtox acute toxicity values, * EC (mg L-1) Daphnia magna acute toxicity values, * EC (mg L-1) 30oC 60oC 30oC 60oC 0. min 150. min 150. min 0. min 150. min 150. min *EC90 *EC *EC *EC50 *EC *EC Raw ww, control 6000 EC40=680 EC30=410 1180 EC30=850 EC25=320 1 h aeration 6000 EC20=563 EC15=358 1180 EC10=197 EC5=119 15 min N2(g) 6000 EC25=456 EC20=456 1180 EC20=164 EC10=148 30 min N2(g) 6000 EC25=445 EC15=442 1180 EC15=142 EC5=120 pH=4.0 6000 EC40=755 EC30=466 1180 EC35=167 EC30=151 pH=7.0 6000 EC40=680 EC30=410 1180 EC30=149 EC25=124 pH=10.0 6000 EC40=653 EC30=465 1180 EC35=210 EC30=182 DO=2 mg L-1 6000 EC35=653 EC25=365 1180 EC20=175 EC15=147 DO=4 mg L-1 6000 EC30=652 EC25=365 1180 EC20=225 EC15=160 DO=6 mg L-1 6000 EC25=652 EC25=364 1180 EC15=190 EC10=132 DO=10 mg L-1 6000 EC20=663 EC20=358 1180 EC15=197 EC5=119 H2O2=100 mg L-1 6000 EC20=653 EC15=664 1180 EC15=170 EC10=135 H2O2=500 mg L-1 6000 EC10=629 EC5=680 1180 EC10=180 EC5=135 H2O2=2000 mg L-1 6000 EC25=650 EC20=664 1180 EC20=123 EC10=113 TiO2=0.1 mg L-1 6000 EC20=655 EC20=738 1180 EC15=200 EC10=260 TiO2=0.5 mg L-1 6000 EC15=652 EC15=665 1180 EC10=115 EC5=105 TiO2=10 mg L-1 6000 EC10=652 EC5=520 1180 EC10=122 EC5=95 TiO2=20 mg L-1 6000 EC25=649 EC20=663 1180 EC20=87 EC15=80 NaCl=1 g L-1 6000 EC15=354 EC10=454 1180 EC15=190 EC10=135 NaCl=2.5 g L-1 6000 EC10=350 EC5=357 1180 EC10=170 EC5=89 NaCl=15 g L-1 6000 EC25=327 EC20=452 1180 EC20=165 EC15=83 Fe+2=2 mg L-1 6000 EC20=353 EC15=354 1180 EC20=125 EC15=110 Fe+2=8 mg L-1 6000 EC15=350 EC10=349 1180 EC15=105 EC10=97 Fe+2=20 mg L-1 6000 EC20=346 EC20=342 1180 EC25=90 EC20=85 Fe+3=10 mg L-1 6000 EC20=353 EC15=354 1180 EC20=160 EC15=130 Fe+3=20 mg L-1 6000 EC15=351 EC10=353 1180 EC15=107 EC10=106 Fe+3=50 mg L-1 6000 EC25=348 EC20=346 1180 EC25=105 EC20=103 HCO3-1=0.5 g L-1 6000 EC25=312 EC20=353 1180 EC25=375 EC15=350 HCO3-1=1 g L-1 6000 EC20=311 EC15=309 1180 EC20=140 EC10=160 HCO3-1=5. g L-1 6000 EC30=308 EC25=308 1180 EC30=170 EC20=162 C4H9OH=0.1 g L-1 6000 EC25=311 EC15=310 1180 EC25=400 EC15=375 C4H9OH=0.5 g L-1 6000 EC20=211 EC10=254 1180 EC20=350 EC10=370 C4H9OH=2 g L-1 6000 EC30=353 EC20=409 1180 EC25=108 EC20=118 * EC values were calculated based on CODdis (mg L-1).
Table 9: Acute toxicity tests (Microtox toxicity and Daphnia magna toxicity) values (EC, mg L-1) of PCI ww before and after sonication process.
Table 9 shows the sensitivity ranking and the EC50 values of PCI ww in Photobacterium phosphoreum and Daphnia magna in sonicated samples containing some additives after 150 min at 60oC. The EC50 value of PCI ww increased from 2.78 mg L-1 (converted from 5 mg L-1 at original EC values in PCI ww for Microtox acute toxicity assay after 150 min) to 25 mg L-1 as the trophic level increased from bacteria in Microtox test to water flea in Daphnia magna test in the sonicated PCI ww samples containing some additives after 150 min at 60oC (Table 9). To verify the relationships between the acute toxicity data of two organisms the EC50 values of Photobacterium phosphoreum were correlated to those of Daphnia magna by statistical analysis using their EC50 values in the effluent of PCI ww. It was found that the EC50 values differed in both test organisms for the sonicated samples containing some additives after 150 min at 60oC. The coefficient of correlation and t-test statistics (R2=0.88, p=0.01, t-test statistics=9.15) show a significant linear relationship between the acute toxicities of PCI ww to Photobacterium phosphoreum and to Daphnia magna in the after 150 min sonication samples The linear corolation was not found between Photobacterium phosphoreum acute toxicity test and Daphnia magna acute toxicity assay (Table 9). This indicated that the water fleas exhibited less acute toxicity to PCI ww. The differences in test sensitivities can be attributed to the differences in the responses of the two different trophic organisms in PCI ww.
The maximum Microtox acute toxicity value decreased from EC90=6000 mg/l to EC10=254 mg L-1 in PCI ww at C4H9OH=0.5 g L-1 and at 60oC after 150 min sonication. The maximum Microtox acute toxicity removal yield was 95.76% in PCI ww at C4H9OH=0.5 g L-1 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity value decreased from EC90=618 mg L-1 to EC15= 80 mg L-1, at TiO2=20 mg L-1 and at 60oC after 150 min sonication. The maximum Daphnia magna acute toxicity removal yield was 93.22% in PCI ww at TiO2=20 mg L-1 and at 60oC after 150 min sonication. As a conclusion, this study showed that the Microtox acute toxicity test is more sensitive than that Daphnia magna in the sonicated PCI ww samples containing some additives after 150 min at 60oC (Table 9).
4.Conclusions
The results of this study showed although sonication alone can be remove the pollutant parameters from PCI industry wastewater, the addition of N2(g), pH, DO, H2O2, TiO2, NaCl, Fe+2, Fe+3 , HCO3-1 and C4H9OH improved the CODdis, TOC and PAHs yields. The maximum CODdis removal yield was 99.68% in PCI ww at 1 h aeration and at NaCl=15 g L-1 and at 60oC after 150 min sonication. The maximum TOC removal yield was 98.35% in PCI ww at DO=10 g L-1 and at 60oC after 150 min sonication. The maximum PAHs removal yield was 99.68% in PCI ww at NaCl=15 g L-1 and at 60oC after 150 min sonication.
This was attributed to the fact that the PAHs, in this study, although less water soluble and non-volatile molecules, tended to accumulate at the gas–liquid interface. Furthermore, it was also found that the concentration of H2O2 formed during PAHs sonication experiments was substantially lower than that formed during water sonication in the absence of PAHs. These PAHs are sonodegraded via pyrolytic reaction that occurs in the hot cavitation bubbles and/or at the gas–liquid interface of the bubbles. The concenration of stronger hydrophobic PAHs increased at the gas–liquid interface of the bubbles with an increase of acidity of wastewater.
The maximum Microtox acute toxicity removal yield was 94.44% in PCI ww at H2O2=500 mg L-1, at TiO2=10 mg L-1, at NaCl=2.5 g L-1 and at 60oC after 150 min sonication, respectively. This study showed that the Microtox acute toxicity test is more sensitive than that Daphnia magna in the sonicated PCI ww samples containing some additives after 150 min at 60oC. The linear corolation was not found between Photobacterium phosphoreum acute toxicity test and Daphnia magna acute toxicity assay.
Acknowledgements
Experimental analyzes in this study were performed at the Laboratories of the Canada Research Center, Ottawa, Canada. The authors would like to thank this body for providing financial support.
References
- Suslick KS, (1989). The chemical effects of ultrasound. Sci Am. 260:80-86.
View at Publisher | View at Google Scholar - Suslick KS, (1989). Ultrasound; its chemical, physical, and biological effects. New York: VCH Publishers.
View at Publisher | View at Google Scholar - Thompson LH and Doraiswamy LK. (1999). Sonochemistry: Science and engineering. Ind Eng Chem Res 38:1215-1249.
View at Publisher | View at Google Scholar - Thompson LH and Doraiswamy LK. (2000). The rate enhancing effect of ultrasound by inducing supersaturation in a solid-liquid system. Chem Eng Sci 55:3085-3090.
View at Publisher | View at Google Scholar - Adewuyi YG. (2001). Sonochemistry: Environmental science and engineering applications. Ind Eng Chem Res. 40:4681-4715.
View at Publisher | View at Google Scholar - Beens J, Blomberg J and Schoenmakers (PJ 2000). Proper tuning of comprehensive two-dimensional gas chromatography (GC×GC) to optimize the separation of complex oil fractions. J High Resolut Chrom. 23:182-188.
View at Publisher | View at Google Scholar - Colussi AJ, Hung HM and Hoffmann MR, Sonochemical degradation rates of contaminants in water. Ultrason Sonochem 3:163-172 (1996).
View at Publisher | View at Google Scholar - Colussi AJ, Hung H-M and Hoffmann MR (1999). Sonochemical degradation rates of volatile solutes. J Phys Chem A. 103:2696-2699.
View at Publisher | View at Google Scholar - David B, Lhote M, Faure V and Boule P. (1998). Ultrasonic and photochemical degradation of chlorpropham and 3-chloroaniline in aqueous solution. Water Res. 32:2451-2461.
View at Publisher | View at Google Scholar - Drijvers D, van Langenhove H and Beckers M. (1999). Decomposition of phenol and trichloroethylene by the ultrasound/H2O2/CuO process. Water Res. 33:1187-1194.
View at Publisher | View at Google Scholar - Kotronarou A, Mills G and Hoffmann MR. (1991). Ultrasonic irradiation of p-Nitrophenol in aqueous solution. J Phys Chem 95:3630-3638.
View at Publisher | View at Google Scholar - Mason TJ, Sonochemistry. (1998). New York: Oxford University Press, New York, USA.
View at Publisher | View at Google Scholar - Mason TJ, Joyce E, Phull SS, Lorimer JP. (2003). Potential uses of ultrasound in the biological decontamination of water. Ultrason Sonochem.10:319-323.
View at Publisher | View at Google Scholar - Okuno H, Yim B, Mizukoshi Y, Nagata Y and Maeda Y, et al. (2000). Sonolytic degradation of hazardous organic compounds in aqueous solution. Ultrason Sonochem 7:261-264.
View at Publisher | View at Google Scholar - Petrier C, Lamy M-F, Francony A, Benahcene A, David B, et al. (1994). Sonochemical degradation of phenol in dilute aqueous solutions: Comparison of the reaction rates at 20 and 487 kHz. J Phys Chem. 98:10514-10520.
View at Publisher | View at Google Scholar - Petrier C, Jiang Y and Iselamy M-F. (1998). Ultrasound and environment: Sonochemical destruction of chloroaromatic derivatives. Environ Sci Technol. 32:1316-1318.
View at Publisher | View at Google Scholar - Psillakis E, Goula G, Kalogerakis N and Mantzavinos D. (2004).Degradation of polcyclic aromatic hydrocarbons in aqueous solutions by ultrasonic irradiation. J Hazard Mater B. 108:95-102.
View at Publisher | View at Google Scholar - Sanchez-Prado L, Barro R, Garcia-Jares C, Liompart M, Lores M, et al. (2008). Sonochemical degradation of triclosan in water and wastewater. Ultrason Sonochem. 15:689-694.
View at Publisher | View at Google Scholar - Serpone N, Terzian R, Hidaka H and Pelizzetti E. (1994). Ultrasonic ınduced dehalogenation and oxidation of 2-, 3-, and 4-chlorophenol in air-equilibrated aqueous media. Similarities with irradiated semiconductor particulates. J Phys Chem. 98:2634-2640.
View at Publisher | View at Google Scholar - Torres RA, Pétrier C, Combet E, Moulet F and Pulgarin C. (2007). Bisphenol A mineralization by integrated ultrasound-UV-iron(II) treatment. Environ Sci Technol. 41:297-302.
View at Publisher | View at Google Scholar - Torres RA, Sarantakos G, Combet E, Pétrier C and Pulgarin C. (2008). Sequential heliophoto-fenton and sonication processes for the treatment of bisphenol A. Photochem Photobiol A Chem. 199:197-203.
View at Publisher | View at Google Scholar - Torres RA, Pétrier C, Combet E, Carrier M and Pulgarin C. (2008). Ultrasonic cavitation applied to the treatment of bisphenol An Effect of sonochemical parameters and analysis of BPA by-products. Ultrason Sonochem 15:605-611.
View at Publisher | View at Google Scholar - Vassilakis C, Pantidou A, Psillakis E, Kalogerakis N and Mantzavinos D. (2004). Sonolysis of natural phenolic compounds in aqueous solutions: degradation pathways and biodegradability. Water Res 38:3110-3118.
View at Publisher | View at Google Scholar - Tiehm A and Neis U. (1999). Ultrasound in Environmental Engineering. Technical University Hamburg-Harburg Reports on Sanitary Engineering. (25), (ISSN 0724-0783, ISBN 3-930400-23-5), GFEUVerlag, Hamburg.
View at Publisher | View at Google Scholar - Tiehm A. (2001). Combination of ultrasonic and biological pollutant elimination. In Mason T and Tiehm A (Eds.), Ultrasound in environmental protection, 6th ed., in Advances in Sonochemistry Elsevier Science, Amsterdam, pp 25-68
View at Publisher | View at Google Scholar - Broman D, Naef C, Rolff C and Zebuehr Y. (1991). Occurrence and dynamics of polychlorinated dibenzo-p-dioxins and dibenzofurans and polycyclic aromatic hydrocarbons in the mixed surface layer of remote coastal and offshore waters of the Baltic. Environ Sci Technol. 25:1850-1864.
View at Publisher | View at Google Scholar - Jones KC, Stratford JA, Waterhouse KS, Furlong ET, Giger W, et al. (1989).Increases in the polynuclear aromatic hydrocarbon content of an agricultural soil over the last century. Environ Sci Technol. 23:95-101
View at Publisher | View at Google Scholar - Marvin CH, McCarry BE, Villella J, Allan LM and Bryant DW. (2000). Chemical and biological process of sediments as indicators of sources of contamination in Hamilton Harbour. Part II: Bioassay-directed fractionation using the Ames Salmonella/microsome assay. Chemosphere. 41:989-999.
View at Publisher | View at Google Scholar - Menzie CA, Potocki BB and Santodonato J. (1992). Exposure to carcinogenic PAHs in the environment. Environ Sci Technol. 26:1278-1284
View at Publisher | View at Google Scholar - Suslick KS. (1986). Organometallic sonochemistry, in advances in organometallic chemistry, in Academic Press, New York, pp 73-119.
View at Publisher | View at Google Scholar - Wheat PE and Tumeo MA. (1997). Ultrasound induced aqueous polycyclic aromatic hydrocarbon reactivity. Ultrason Sonochem. 4:55-59
View at Publisher | View at Google Scholar - Cataldo F. (2000). Ultrasound-induced cracking and pyrolysis of some aromatic and naphthetic hydrocarbons. Ultrason Sonochem .7:35-43.
View at Publisher | View at Google Scholar - David B and Riguier D. (2000). Ultrasonication of aqueous and micellar suspensions of anthracene fixed on silica. Ultrason Sonochem. 9:45-52.
View at Publisher | View at Google Scholar - Laughrey Z, Bear E, Jones R and Tarr MA. (2001). Aqueous sonolytic decomposition of polycyclic aromatic hydrocarbons in the presence of additional dissolved species. Ultrason Sonochem. 8:353-357.
View at Publisher | View at Google Scholar - Little C, Hepher MJ and El-Sharif M. (2002). The sono-degradation of phenanthrene in an aqueous environment. Ultrasonics. 40:667-674.
View at Publisher | View at Google Scholar - Psillakis E, Ntelekos A, Mantzavinos D, Nikolopoulos E and Kalogerakis N. (2003). Solid-phase microextraction to monitor the sonochemical degradation of polycyclic aromatic hydrocarbons in water. J Environ Monitor. 5:135-140.
View at Publisher | View at Google Scholar - Taylor Jr. E, Cook BB and Tarr MA. (1999). Dissolved organic matter inhibition of sonochemical degradation of aqueous polycyclic aromatic hydrocarbons. Ultrason Sonochem. 6:175-183.
View at Publisher | View at Google Scholar - Banjoo DR and Nelson PK. (2005). Improved ultrasonic extraction procedure for the determination of polycyclic aromatic hydrocarbons in sediments. J Chromatogr A. 1066:9-18.
View at Publisher | View at Google Scholar - Callahan MW, Slimak NW, Gabelc IP, May CF, Fowler JR, et al. (1979). Washington DC: EPA-440/4-79-029, U.S. EPA.
View at Publisher | View at Google Scholar - (1994). European Union’s European Economic Community (EU EEC) Directive 1994. Journal officiel des Communautés Européennes. N 80/778, July 15, 1980; N 76/464, May 18, 1990 and N 67/548, 1994.
View at Publisher | View at Google Scholar - (2006). United States Environmental Protection Agency (U.S. EPA) & Oregon Department of Environmental Quality [DEQ], Portland harbor joint source control strategy, in Final Report, 2009.
View at Publisher | View at Google Scholar - Kim Oanh NT, Nghiem LH and Phyu YL. (2002). Emission of polycyclic aromatic hydrocarbons, toxicity and mutagenicity from domestic cooking using sawdust briquettes, wood and kerosene. Environ Sci Technol. 36:833-839.
View at Publisher | View at Google Scholar - Papadaki M, Emery RJ, Abu-Hassan MA, Diaz-Bustos A, Metcalfe IS and Mantzavinos D. (2002). Sonocatalytic oxidation processes for the removal of contaminants containing aromatic rings from aqueous effluents. Sep Purif. 34:35-42.
View at Publisher | View at Google Scholar - Huang W, Tang X, Felner I, Koltypin Y and Gedanken A. (2004). Preparation and characterization of FexOy – TiO2 via sonochemical synthesis. Mater Res Bull. 37:1721-1735.
View at Publisher | View at Google Scholar - Suslick KS. (2000). Sonoluminescence and sonochemistry, in Philos Trans R Soc Lond A 361:342-368
View at Publisher | View at Google Scholar - Lipps, W.C., Braun-Howland, E.B. and Baxter, T.E. (2022). Standard Methods for the Examination of Water and Wastewater. 24th. Edition, Lipps, W.C., Braun-Howland, E.B. and Baxter, T.E. (editors), American Public Health Association (APHA), American Water Works Association (AWWA), Water Environment Federation (WEF), Elevate Your Standards. American Public Health Association 800 I Street, NW Washington DC: 20001-3770, USA.
View at Publisher | View at Google Scholar - (1993). Microtox acute toxicity test, DIN 38412 L34, L341.
View at Publisher | View at Google Scholar - Lange B. LUMISmini, (1994). Operating Manual. Dr. Bruno Lange, Düsseldorf, Germany,
View at Publisher | View at Google Scholar - Zar JH. (1984). Biostatistical analysis, 2nd. Ed., New Jersey: Prentice Hall, Englewood Cliffs., pp 718-736.
View at Publisher | View at Google Scholar - (2005). STATGRAPHICS Centurion XV, software, 2005 (Statpoint, Inc). StatPoint, Inc. Statgraphics Centurion XV, Herndon, VA. USA.
View at Publisher | View at Google Scholar - Kojima Y, Fujikta T, Ona EP, Matsuda H, Koda S, et. al. (2005). Effect of dissolved gas species on ultrasonic degradation of (4-chloro-2-methylphenoxy) acetic acid (MCPA) in aqueous solution. Ultrason Sonochem. 12:359-365.
View at Publisher | View at Google Scholar - Suslick KS, in Suslick K. S. (Ed.). (1988). In Ultrasound: Its chemical, physical and biological effects, VCH Publisher., pp 129.
View at Publisher | View at Google Scholar - Ince NH, Gultekin I and Tezcanli-Guyer G. (2009). Sonochemical destruction of nonylphenol: effects of pH and hydroxyl radical scavengers. J Hazard Mater. 172:739-743.
View at Publisher | View at Google Scholar - Lawton E and Show D. (2009). Sonication of pulp and paper effluent. Ultrason Sonochem. 16:321-324.
View at Publisher | View at Google Scholar - Wu Z and Ondruschka B. (2005). Roles hydrophobicity and volatility of organic substrates on sonolytic kinetics in aqueous solutions. J Phys Chem. 109:6521-6526.
View at Publisher | View at Google Scholar - David B. (2009). Sonochemical degradation of PAH in aqueous solution. Part I: Monocomponent PAH solution. Ultrason Sonochem. 16:260-265.
View at Publisher | View at Google Scholar - Mendez-Arriaga F, Esplugas S and Jimenez J. (2008). Photocatalytic degradation of non-steroidal anti-inflammatory drugs with TiO2 and simulated solar irradiation. Water Res. 42:585-594.
View at Publisher | View at Google Scholar - Sivasankar T and Moholkar VS. (2009). Physical insights into the sonochemical degradation of recalcitrant organic pollutants with cavitation bubble dynamics. Ultrason Sonochem. 16:769-781.
View at Publisher | View at Google Scholar - Goskonda S, Catallo WJ and Junk T. (2002). Sonochemical degradation of aromatic organic pollutants. Waste Manage. 22:351-356.
View at Publisher | View at Google Scholar - Naddeo V, Belgiorno V and Napoli RMA. (2007). Behaviour of natural organic matter during ultrasonic irradiation. Desalination. 210:175-182.
View at Publisher | View at Google Scholar - Naddeo V, Belgiorno V, Kassinos D, Mantzavinos D and Meric S. et al. (2010). Ultrasonic degradation, mineralization and detoxification of diclofenac in water: Optimization of operating parameters. Ultrason Sonochem. 17:179-185.
View at Publisher | View at Google Scholar - Olson TM and Barbier PF. (1994).Oxidation kinetics of natural organic matter by sonolysis and ozone. Water Res. 28:1383-139.
View at Publisher | View at Google Scholar - Tuziuti T, Yasui K, Lee J, Kozuka T, Towata A et al. (2009). Influence of surface active solute on ultrasonic waveform distortion in liquid containing air bubbles. J Phys Chem A. 31:8893-8900.
View at Publisher | View at Google Scholar - Yim B, Okuno H, Nagata Y and Maeda Y. (2001). Sonochemical degradation of chlorinated hydrocarbons using a batch and continuous flow system. J Hazard Mater B. 81:253-263.
View at Publisher | View at Google Scholar - Yim B, Nagata Y and Maeda Y, Sonolytic degradation of phthalic acid esters in aqueous solutions. Acceleration of hydrolysis by sonochemical action. J Phys Chem A. 106:104-107.
View at Publisher | View at Google Scholar - Yim B, Yoo Y and Maeda Y. (2002). Sonolysis of alkyphenols in aqueous solution with Fe (II) and Fe (III). Chemosphere. 50:1015-1023 (2003).
View at Publisher | View at Google Scholar - Park JK, Hong SW and Chang WS. (2000). Degradation of polycyclic aromatic hydrocarbons by ultrasonic irradiation. Environ Technol 21:1317-1323.
View at Publisher | View at Google Scholar - Gogate PR and Pandit AB. (2004). A review of imperative technologies for wastewater treatment II: hybrid methods. Adv Environ Res. 8:553-597.
View at Publisher | View at Google Scholar - Lindsey ME and Tarr MA. (2000). Quantitation of hydroxyl radical during fenton oxidation following a single addition of iron and peroxide. Chemosphere. 41:409-417.
View at Publisher | View at Google Scholar - Wen S, Zhao J, Sheng G, Fu J and Peng P. (2003). Photocatalytic reactions of pyrene at TiO2/water interfaces. Chemosphere. 50:111-119.
View at Publisher | View at Google Scholar - Hung HM and Hoffmann MR. (1998). Kinetics and mechanism of the enhanced reductive degradation of CCl4 by elemental iron in the presence of ultrasound. Environ Sci Technol 32:3011-3016.
View at Publisher | View at Google Scholar - Chakinala AG, Gogate PR, Burgess AE and Bremner DH. (2008). Treatment of industrial wastewater effluent using hydrodynamic cavitaation and the advanced fenton process. Ultrason Sonochem. 15:49-54.
View at Publisher | View at Google Scholar - Rae J, Ashokkumar M, Eulaerts O, Von Sonntag C, Reisse J et al. (2005). Estimation of ultrasound induced cavitation bubble temperatures in aqueous solutions. Ultrason Sonochem. 12:325-329.
View at Publisher | View at Google Scholar - Benabdallah El-Hadj T, Dosta J, Marquez-Serrano R and Mata-Alvarez J. (2007). Effect of ultrasound pretreatment in mesophilic and thermophilic anaerobic digestion with emphasis on naphthalene and pyrene removal. Water Res. 41:87-94.
View at Publisher | View at Google Scholar - Lindsey ME and Tarr MA. (2000). Inhibition of hydroxyl radical reaction with aromatics by dissolved natural organic matter. Environ Sci Technol. 34:444-449.
View at Publisher | View at Google Scholar - Wang J, Ma T, Zhang Z, Zhang X, Jiang Y, et al. (2006). Investigation on the sonocatalytic degradation of parathion in the presence of nanometer rutile titanium dioxide (TiO2) catalysis. J Hazard Mater B. 137:972-980.
View at Publisher | View at Google Scholar - Liu G, Li X, Zhao J, Hidaka H and Serpone N. (2000). Photooxidation pathway of sulforhodamine-B. Dependence on the adsorption mode on TiO2 exposed to visible light radiation. Environ Sci Technol. 34:3982-4000.
View at Publisher | View at Google Scholar - Wang J, Sun W, Zhang Z, Jiang Z, Wang X, et al. (2008). Preparation of Fe-doped mixed crystal TiO2 catalyst and investigation of its sonocatalytic activity during degradation of azo funchsine under ultrasonic irradiation. J Colloid Interface Sci. 320:202-209.
View at Publisher | View at Google Scholar - Sivasankar T and Moholkar VS. (2008). Physical features of sonochemical degradation of nitroaromatic pollutants. Chemosphere. 72:1795-1806.
View at Publisher | View at Google Scholar - Sunartio D, Ashokkumar M and Grieser F, (2007). Study of the coalescence of acoustic bubbles as a function of frequency, power, and water-soluble additives. J Am Chem Soc. 129:6031-6036.
View at Publisher | View at Google Scholar - Bremmer DH, Di Carlo S, Chakinala AG and Cravotto G. (2008). Mineralisation of 2,4-dichloropheoxyacetic acid by acoustic or hydrodynamic cavitation in conjunction with the advaced fenton process. Ultrason Sonochem. 15:416-419.
View at Publisher | View at Google Scholar - Beckett MA and Hua I. (2000). Elucidation of the 1,4-dioxane decomposition pathway at discrete ultrasonic frequencies. Environ Sci Technol. 34:3944-3953.
View at Publisher | View at Google Scholar - Beckett MA and Hua I. (2003). Enhanced sonochemical decomposition of 1,4-dioxane by ferrous iron. Water Res. 37:2372-2376.
View at Publisher | View at Google Scholar - Guo L, Li X-M, Bo X, Yang Q, Zeng G-M, et al. (2008). Impacts of sterilization, microwave and ultrasonication pretreatment on hydrogen producing using waste sludge. Bioresour Technol. 99:3651-3658.
View at Publisher | View at Google Scholar - Wang J, Pan Z, Zhang Z, Zhang X, Wen F, et al. (2006). Sonocatalytic degradation of methyl parathion in the presence of nanometer and ordinary anatase titanium dioxide catalysts and comparison of their sonocatalytic abilities. Ultrason Sonochem. 13:493-500.
View at Publisher | View at Google Scholar - Son H-S, Choi S-B, Khan E and Zoh K-D. (2006). Removal of 1,4-dioxane from water using sonication: Effect of adding oxidants on the degradation kinetics. Water Res. 40:692-698.
View at Publisher | View at Google Scholar - Shaw LE and Lee D. (2009). Sonication of pulp and paper effluent. Ultrason Sonochem. 16:321-324.
View at Publisher | View at Google Scholar - Suslick KS, Gaurenoski JJ, Schubert PF and Wang HH. (1984). Sonochemistry an non-aqueous solutions. Ultrasonics. 22:23-36.
View at Publisher | View at Google Scholar - Wayment DG and Casadonte Jr. DJ. (2002). Frequency effect on the sonochemistry remediation of alachlor, Ultrason Sonochem. 9:251-257.
View at Publisher | View at Google Scholar - Quesada-Penate I, Julcour-Lebigue C, Jauregui-Haza U-J, Wilhelm A-M and Darie DH. (2009).Sonolysis of levodopa and paracetamol in aqueous solutions. Ultrason Sonochem. 16:610-616.
View at Publisher | View at Google Scholar - Tauber A, Mark G, Schuchmann H and von Sonntag C. (1999). Sonolysis of tert-butyl alcohol in aqueous systems. Ultrason Sonochem. 9:291-296.
View at Publisher | View at Google Scholar - Tauber A, Mark G, Schuchmann H-P and von Sonntag C. (1999b) Sonolysis of tert-butyl alcohol in aqueous solution. J Chem Soc Perkin Trans. 2:1129-1135.
View at Publisher | View at Google Scholar - Grcic I, Dinko V and Natalija N. (2010). Modeling the mineralization and discoloration in colored systems by (US)Fe+2/H2O2/S2O8−2 processes: A proposed degradation pathway. Chem Eng. 157:35-44.
View at Publisher | View at Google Scholar - Chakinala AG, Gogate PR, Chand R, Bremner DH, Molina R et al. (2008). Intensification of oxidation capacity using chloroalkanes as additives in hydrodynamic and acoustic cavitation reactors. Ultrason Sonochem. 15:164-170.
View at Publisher | View at Google Scholar - Sponza DT. (2001). Assessment of toxicity in paper mill and chemical industry wastewaters. Biyoteknoloji Dergisi. 25:41-55.
View at Publisher | View at Google Scholar - Sponza DT. (2001). Toxicity survey in the effluents of leather, textile and petrochemical industry treatment plants. SKKD. 11:17-30.
View at Publisher | View at Google Scholar - Sponza DT. (2002). Incorporation of toxicity tests to the discharges of pulp and paper industry in Turkey. Bull Environ Contam Toxicol. 69:719-726.
View at Publisher | View at Google Scholar - Sponza DT. (2002). Necessity of toxicity assessment in Turkish industrial discharges (examples from metal and textile industry effluents). Environ Monit Assess. 73:41-66.
View at Publisher | View at Google Scholar - Sponza DT. (2003). Application of toxicity tests into discharges of the pulp-paper industry in Turkey. Ecotox Environ Safe. 54:74-86.
View at Publisher | View at Google Scholar - Sponza DT. (2006). Toxicity studies in a chemical dye production industry in Turkey. J Hazard Mater A. 138:438-447.
View at Publisher | View at Google Scholar - Sponza DT, Selcuk-Kuscu O. (2011). Relationships between acute toxicities of paranitrophenol (p-NP) and nitrobenzene (NB) to Daphnia magna and Photobacterium phosphoreum: Physicochemical properties and metabolites under anaerobic/aerobic sequentials. J Hazard Mater. 185:1187-1197.
View at Publisher | View at Google Scholar
