

Research Article | DOI: https://doi.org/10.31579/2834-5118/014
New Reports of Col1a2, Col9a2, Tp63, And Dhdds Gene Mutations Associated with Skeletal Abnormalities with Focus on Osteogenesis Imperfecta
1 Department of Obstetrics and Gynecology, Clinical Research Development Center, Emam Reza hospital, Kermanshah University of Medical Sciences, Kermanshah, Iran.University Of North Carolina at Greensboro, USA
2 Medical Genetic Lab, Kermanshah, Iran. North Central University, CA Iran.
3 Division of Genetics, Department of Cell and Molecular Biology and Microbiology, Faculty of Science and Biotechnology, University of Isfahan, Isfahan, IR Iran.
4 Biology Department, College of Bioscience, Islamic Azad University, Tehran North Branch, Tehran, Iran.
*Corresponding Author: Vahid Omarmeli, Biology Department, College of Bioscience, Islamic Azad University, Tehran North Branch, Tehran, Iran.
Citation: Nasrin Mansouri, Marjan Assefi, Sohila Nankali, Alireza Sharafshah, Vahid Omarmeli. (2023), New reports of COL1A2, COL9A2, TP63, and DHDDS gene mutations associated with skeletal abnormalities with focus on Osteogenesis Imperfecta. International Journal of Clinical Surgery, 2(1); DOI:10.31579/2834-5118/014
Copyright: © 2023 Vahid Omarmeli, this is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 15 November 2022 | Accepted: 26 December 2022 | Published: 09 January 2023
Keywords:
Abstract
Objective: Osteogenesis imperfecta (OI) as an inherited disorder is an important disease that should be screened before pregnancy. The type of OI inheritance includes types I to V with the autosomal dominant model of inheritance and most often results from new dominant mutations. This pattern of inheritance is responsible for 90 to 95% of cases. There are shreds of evidence for germ cell mosaics as a justification for having more than one child with the disease in families with healthy parents.
Methods: After genetic counseling, Whole Exome Sequencing (WES) technique was used to look for plausible pathogenic mutations in the aborted fetus. Also, candidate mutations were examined in her parents by sanger sequencing. For further investigations, in silico analyses were perfumed through I-TASSER, SWISS-MODEL, MolProbity, ProSA, PyMol, and TFBIND tools.
Results: Four mutations were found in aborted fetus including c.1081G>A: p.Gly361Ser (COL1A2), c.226G>A: p.Asp76Asn (TP63), and c.281T>C: p.Met94Thr (DHDDS), and c.629C>T: p.Pro210Leu (COL9A2). COL1A2 mutation was new in the fetus and other mutant alleles of the three remaining mutations were inherited from one of the parents.
Conclusion: In conclusion, in vitro, and in silico analysis suggested that this study is the first report to indicate the epistasis effect of COL1A2 and COL9A2 missense mutations for a severe pathogenic impact leads to OI type II and lethality of pregnancies.
Introduction
As a systemic connective tissue disorder, Osteogenesis imperfecta (OI) is discriminated through low bone mass and fragility which produces significant morbidity because of pain, immobility, skeletal malformations, and stunted growth [1, 2]. Reduced bone strength leads to minimally traumatic breaks or breaks in atypical positions [2]. Screening of variants in OI-associated genes in lightly affected or obviously asymptomatic people confirms the presence of a wider clinical spectrum, ranging from low bone mass and early onset osteoporosis to progressively deforming and lethal perinatal OI [2]. There are two genotype-phenotype associations described in OMIM for Osteogenesis imperfecta type II (OI2). OI2 is produced by heterozygous mutation in the COL1A2 gene or in the COL1A1 gene at 7q21.3 and 17q21.33, respectively. An autosomal dominant model of inheritance is suggested for OI2. The most severely affected cases are born from non-affected parents. This phenomenon might be due to parental gonadal mosaicism or novel mutations [3].
Ectrodactyly-ectodermal cleft syndrome 3 (EEC) stands as one of six overlapping syndromes produced by mutations in the tumor protein p63 gene (TP63) [4]. EEC is frequently suspected when patients present with a compound of cleft hands or feet, polydactyly and syndactyly, abnormal development of structures derived from the ectoderm, including teeth, hair, skin, and nails, and cleft lip and palate [5]. Also, mutations in DHDDS gene are connected to developmental delay and seizures with or without movement abnormalities [6].
Here, by WES screening the aborted fetus and sanger sequencing of her parents, we report two novel mutations (Asp76Asn mutation in TP63 gene and Met94Thr DHDDS gene) and two mutations with vital impacts on morbidity of fetus associated with OI (Pro210Leu in COL1A2 gene and Pro210Leu in COL9A2 gene).
Case Presentation
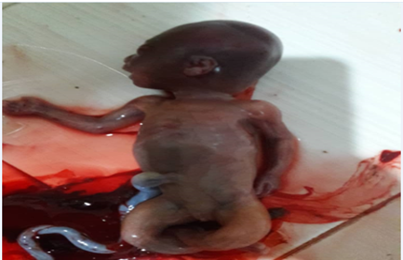
An aborted female fetus was investigated for putative mutations by WES. To find the cause of miscarriage, her parents were under investigation of molecular genetic tests, too. The parents had no familial history. Pregnancy with a live fetus was observed with a floating and breech display. Fetal activity, movements, and heart rate were normal (FHR = 158 bpm). Gestational age based on fetal biometric factors including BPD, HC, AC, and OFD was about 18w + 1d and according to HL, FL was about 16w which was indicative of Proximal Significant shortness of both lower and upper limbs. The femur on both sides was associated with deformity and bowing, and the FL / Foot ratio was about 70% and out of the standard range due to gestational age. Based on the above evidence, the possibility of skeletal dysplasia is strongly raised. The pair was in the anterior position of grade 0-I. The maximum thickness of the pair was 21 mm and the distance between the pair and the IO was about 28 mm. The volume of normal amniotic fluid (AFI = 12.8cm) and the thickness of her largest sac was about 3.4cm. Fetal weight based on biometric factors was about 186gr ±10%. The length of the cervical canal was about 38 mm and the entrance to the cervical canal was closed. Cervical canal length is important considering clinical findings and gestational age. Examination of 4chamber calvary, spine, heart brain, kidneys, diaphragm, abdominal wall to an extent that could be examined by ultrasound showed no evidence of anomaly. The symptoms of the aborted fetus were completely matched with OI symptoms included short and fractured limbs, thin chest with hypomineralization and several fractures in ribs and long bones (Figure 1).
Material and Methods
In vitro investigations
WES was utilized to detect the putative pathogenic mutations in patient as follows: Genomic deoxyribonucleic acid (gDNA) was isolated from the patient’s specimen using a filter-based methodology and quantified. A total amount of 1.0μg genomic DNA per sample was used as input material for the DNA sample preparation. Sequencing libraries were generated using Agilent SureSelect Human All ExonV7 kit (Agilent Technologies, CA, USA) following manufacturer’s recommendations and x index codes were added to attribute sequences to sample. Briefly, fragmentations were carried out by hydrodynamic shearing system (Covaris, Massachusetts, USA) to generate 180-280bp fragments. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities and enzymes were removed. After adenylation of 3’ ends of DNA fragments, adapter oligonucleotides were ligated. DNA fragments with ligated adapter molecules on both ends were selectively enriched in a PCR reaction. Captured libraries were enriched in a PCR reaction to add index tags to prepare for hybridization. Products were purified using AMPure XP system (Beckman Coulter, Beverly, USA) and quantified using the Agilent high sensitivity DNA assay on the Agilent Bioanalyzer 2100 system. The qualified libraries are fed into NovaSeq 6000 Illumina sequencers. Then data quality control, analysis and interpretation were run on G9 generation of HP server using unix based operating system.
In silico investigations
For further findings, the current report designed and done the homology modeling and superimposition evaluations for COL1A2, COL9A2, p63, and DHDDS protein structures. Homology Modeling was done through I-TASSER, SWISS MODEL, and PHYRE2 and also, evaluated by Molprobity and ProSA online software (available at https://prosa.services.came.sbg.ac.at/prosa.php and https://prosa.services.came.sbg.ac.at/prosa.php ). Final models were visualized by the PyMOL Molecular Graphics System (Version 2.0 Schrödinger, LLC) and also FATCAT (available at https://fatcat.godziklab.org/ ). TFBIND (available at: https://tfbind.hgc.jp/ ) was used for the prediction of probable alterations in transcription factor binding affinity.
Results
Molecular reports
During the study performed on the exom of the aborted fetus, and due to the reason of referral (unrelated marriage and having a history of 2 abortion treatments with severe skeletal abnormalities), notable variations were seen in important genes. Due to the limitations of Exome 4 Variant test with autosomal dominant pattern which was observed as Pathogenic, Likely Pathogenic and Uncertain Significance in his aborted fetal sample. Confirmation of mutation on variants showed that mutation in COL1A2 gene is new and parents do not have this mutation. This mutation will cause OI type 2, which is consistent with clinical observations on the embryonic specimen. Also, due to the recurrence of this abnormality (2 affected fetuses), a mutation in the sex cell line has apparently occurred. Other mutations in COL9A2, TP53, and DHDDS genes were listed in Table 1.
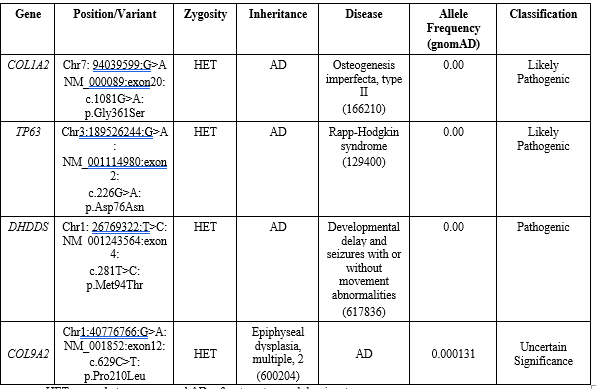
HET means heterozygous and AD refers to autosomal dominant.
Bioinformatics report
The modeled structures were finalized in both wild-type and mutant alleles for every four proteins reported from WES test. At first step, MuPro online server predicted both value and sign of energy change using SVM and sequence information (delta delta G=-0.55) which showed decrease stability in the mutant structure. The models of I-TASSER, SWISS-MODEL, and PHYRE2 were not proper enough to report (Figures are not shown).
Regulatory analysis showed rs199831035 (Gly351Ser) is located in CTCF regulatory site of COL1A2 gene. Further investigations revealed that by changing nucleotide G to A, some transcription factors (TF) can not bind to this region and new TF binding site cause the binding site for new TFs. TFs including v-MYB, NF KAPPA B50, NF KAPPA B65, and p53 just bind when G allele is present in the sequence region of 30 nucleotides (>1 dna: chromosome: GRCh38:1:40311080:40311111:1 CCCTTGCACTCACCGGCTTCCCCTGGTGGCCA) and YY1, ZID, STAF, E47, MYOD, LMO2COM, and CETS1P54 when A allele is in the aforementioned sequence. This analysis was performed for rs199831035 (Pro210Leu). Locating in a flanking promoter region, the substitution of G to A allele leads to disruption/formation TF binding sites including SP1, PAX2, GC, MZF1, NFKAPPAB50 which disrupt in A form and N-MYC, P300, EVI1, and IRF2 new binding sites in A allele presence.
Discussion
Our results revealed that the aborted fetus was heterozygote for every for reported mutations; however, one of her parents had the mutant allele except for COL1A2 gene. Both of parents were homozygotes for G allele and aborted fetus showed new mutation (G/A). Here, we found certain lethality by epistasis effects of c.1081G>A COL1A2 and c.629C>T COL9A1 gene mutations causing OI type II.
As far as we know, there is only a report for Gly361Ser by Talebi et al in an Iranian family. They found Gly361Ser for the first time in two individuals as heterozygotes. By NGS analysis, they found COL1A1 and COL1A2 gene mutations [7]. The Glysine to Serine change at the Gly position of a Gly-X-Y repeat consequences in the loss of non-polar side chains and alteration with residues having polar sidechains. This replacement is expected to negatively affect the role of the COL1A2 protein through disrupting her uninterrupted Gly-X-Y triplets. Previous reports stated that the substitution of glycine (smallest amino acid) with other amino acids for instance Arg, Ala, serine, or Cys damages the stability and proper folding of the collagen triple helix, the integration of mature collagen into the extracellular matrix, the secretion of procollagen from the cell, and extracellular processing levels [8].
We suggest that the reason of our case’s miscarriage can be the epistasis impact of having two mutations in COL1A2 and COL9A2 genes; more specifically, none of the case’s parents were heterozygotes for both of these genes. The main cause of this phenomenon might be due to the regulation alterations in these two mutations. In silico investigations represented that rs1791894410 (Gly361Ser) and rs199831035 (Pro210Leu) are in promoter flanking and CTCF regions, respectively. Gly361Ser site showed two important changes in TF affinity including p53 disruption in mutant allele and formation of YY1 binding site in mutant form. Dual role of p53 is reported in animal studies showing its vital role in osteoblast differentiation and survival. p53-deficient studies on mice showed elevated bone mass and bone formation rates, probable through improved osteoblast proliferation and decreased apoptosis [9]. Interestingly, p53 acts as a modulator of the COL1A2 gene. Genes involved in p53-dependent stress response were investigated in normal cells and tissues which were irradiated versus a background of differential p53 expression. In the case of gamma-irradiated fibroblasts, Komarova et. al confirmed that COL1A2 is upregulated in a p53-dependent mode and acts as a growth repressor [10]. In fibroblasts, YY1 binding sites are essential for strong transcription directed by the Col1a1 core promoter [11]. This proposes that its function is separate from the inhibitory role in osteoblasts played by this TF in vitamin D activation of the osteocalcin gene [12].
Pro210Leu site bioinformatics prediction indicated that the most important TF SP1 site will disrupt in A allele. Abundant studies documented the SP1 role in association with bone mineral density and osteoporotic fracture [13, 14]. Another study found that Sp1/Sp3 and MZF1 are vital TFs regulating N-cadherin promoter activity and expression in osteoblasts [15]. Evi1 was one of the TFs that its biding site will form by A allele presence. There are evidences for Evi1 role in bone marrow failure, erythroid dysplasia, and increased apoptosis [16].
Conclusively, due to the dominant pattern of COL1A2 gene and the disease, the probability of future pregnancies of this couple is 50% and PND is strongly recommended in every pregnancy. Fortunately, the parents have 3 other variants, which do not threaten future pregnancies due to the health of the couple. For the first time, this study suggested the epistasis effects of two critical genes involved in OI disease (COL1A2 and COL9A2) leading to miscarriage and lethality for fetus.
Acknowledgements
This an honor to thank from all of our colleagues in the Dr. Shaveisi zadeh lab for their sincere and compassionate cooperation.
Conflict of Interest
The authors have no conflict of interest to disclose.
Authors contribution
Vahid Omarmeli did the main designing, distinguishing the clinical steps. Masoumeh Favaedi, Hanieh Faizmahdavi, and Parichehr Darabi contributed in the paper preparation. Soheila Nankali and Marjan Assefi overviewed the case study section. Alireza Sharafshah and Zhila Shaveisi-zadeh contributed in the paper writing and finally, Nasrin Mansouri supervised the whole project and contributed in all parts.
References
- Marini, J.C., et al. (2017). Osteogenesis imperfecta. Nature reviews Disease primers. 3(1): p. 1-19.
View at Publisher | View at Google Scholar - Robinson, M.-E. and F. (2019). Rauch, Mendelian bone fragility disorders. Bone. 126: 11-17.
View at Publisher | View at Google Scholar - Byers, P., et al. (1988). Perinatal lethal osteogenesis imperfecta (OI type II): a biochemically heterogeneous disorder usually due to new mutations in the genes for type I collagen. American journal of human genetics. 42(2): p. 237.
View at Publisher | View at Google Scholar - Rinne, T., et al. (2006). Pattern of p63 mutations and their phenotypes—update. American Journal of Medical Genetics Part A. 140(13): 1396-1406.
View at Publisher | View at Google Scholar - Rinne, T., H.G. (2007). Brunner, and H. van Bokhoven, p63-associated disorders. Cell cycle, 6(3): p. 262-268.
View at Publisher | View at Google Scholar - Hamdan, F.F., et al. (2017). High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. The American Journal of Human Genetics. 101(5): p. 664-685.
View at Publisher | View at Google Scholar - Talebi, F., et al. (2017). Next-generation sequencing reveals one novel missense mutation in COL1A2 gene in an Iranian family with osteogenesis imperfecta. Iranian biomedical journal. 21(5): p. 338.
View at Publisher | View at Google Scholar - Byers, P. and W. (1993). Cole, Connective tissue and its heritable disorders. Molecular, genetic and medical aspects (ed. PM Royce and B. Steinmann). p. 317-350.
View at Publisher | View at Google Scholar - Wang, X., et al. (2006). p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. The Journal of cell biology. 172(1), 115-125.
View at Publisher | View at Google Scholar - Komori, T. (2016). Cell death in chondrocytes, osteoblasts, and osteocytes. International journal of molecular sciences. 17(12): 2045.
View at Publisher | View at Google Scholar - Riquet, F.B., et al. (2001). YY1 is a positive regulator of transcription of theCol1a1 gene. Journal of Biological Chemistry. 276(42): p. 38665-38672.
View at Publisher | View at Google Scholar - Guo, B., et al. (1997). YY1 regulates vitamin D receptor/retinoid X receptor mediated transactivation of the vitamin D responsive osteocalcin gene. Proceedings of the National Academy of Sciences. 94(1): 121-126.
View at Publisher | View at Google Scholar - Mann, V. and S. (2003). Ralston, Meta-analysis of COL1A1 Sp1 polymorphism in relation to bone mineral density and osteoporotic fracture. Bone. 32(6): p. 711-717.
View at Publisher | View at Google Scholar - Khoschnau, S., et al. (2008). Type I collagen α1 Sp1 polymorphism and the risk of cruciate ligament ruptures or shoulder dislocations. The American journal of sports medicine. 36(12): 2432-2436.
View at Publisher | View at Google Scholar - Le Mee, S., O. Fromigue, and P. (2005). Marie, Sp1/Sp3 and the myeloid zinc finger gene MZF1 regulate the human N-cadherin promoter in osteoblasts. Experimental cell research. 302(1): 129-142.
View at Publisher | View at Google Scholar - Nilsson, S.K., et al. (1999). Cells capable of bone production engraft from whole bone marrow transplants in nonablated mice. The Journal of experimental medicine, 189(4): p. 729-734.
View at Publisher | View at Google Scholar
