

Case report | DOI: https://doi.org/10.31579/2834-8508/034
Neuroendocrine Tumor of the Appendix: A Case Report and Review of Literature
*Corresponding Author: Nabil Chettahi, Pediatric Surgery Department – Hassan II University Hospital, Fez, Morocco
Citation: Nabil Chettahi, Abdelhalim Mahmoudi, Othmane Alaoui, Khalid Khattala, Youssef Bouabdallah (2024), Neuroendocrine Tumor of the Appendix: A Case Report and Review of Literature. Archives of Clinical and Experimental Pathology. 3(5); Doi:10.31579/2834-8508/034
Copyright: © 2024 Nabil Chettahi, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 06 August 2024 | Accepted: 23 August 2024 | Published: 04 September 2024
Keywords: appendix; appendectomy; case report; neuroendocrine tumor; neoplasms; cell proliferation.
Abstract
Background: Neuroendocrine tumor of the appendix is a rare neoplasm discovered only incidentally. They are mainly found in the histological examination of appendices operated due to acute appendicitis. The treatment is essentially surgical, and depending on the size of the lesion, it can range from a simple appendectomy to right hemicolectomy.
Case presentation: In our work, we report the case of a neuroendocrine tumor of the appendix in a 15- year-old female patient admitted for symptoms of acute abdominal pain. She was diagnosed with appendicular peritonitis and admitted to the surgical unit. The anatomopathological examination of the vermiform appendix obtained after appendicectomy showed a histological aspect of a neuroendocrine tumor.
Conclusion: We recommend that appendectomy specimens be routinely sent for pathological examination malignancy and NETs, may be missed without histopathological examination.
Background
Neuroendocrine tumors [NETs] are rare tumors, representing less than 1% of all malignant tumors, and more than 67.5% of them occur in the digestive tract [1]. The appendicular location was described for the first time in 1914 by Gosset and Masson [2]. The vast majority of them are located at the tip of the appendix and remain, therefore, generally asymptomatic.
The diagnosis of certainty remains histological. It allows specifying the mitotic index, the proliferation index and thus classifies NETs according to their degree of differentiation [3].
In this study, we report the case of a primary neuroendocrine tumor of the appendix.
Case Presentation
A 15-year-old female patient, with no previous pathological history, was admitted to the pediatric emergency department of the Hassan II inter-university hospital of Fez, for generalized abdominal pain.
The history of her illness dates back to 8 days before her admission with the onset of generalized abdominal pain, accompanied by food vomiting without transit disorders, evolving in a context of fever and alteration of the general state.
On her admission, the patient was conscious, in fairly good general condition, febrile at 39 °C, tachycardia, with diffuse abdominal guarding.
A workup was ordered, showing:
• CBC-platelets: WBC: 7000 / Hb: 8.3
• C-reactive protein: 225
• Cytobacteriological examination of urine (CBEU): leucocyturia: 25200.
• The abdominal echography showed the presence of a blind digestive structure appended to the cecum, non-peristaltic, painful under the probe and measuring 8 mm at its tip.
The distal extremity of the appendix is in contact with a collection of fine echogenic content fusing along the right flank and subhepatically + intraperitoneal effusion blade at the RIF (Right Iliac Fossa).
Based on these findings, the patient was diagnosed with appendicular peritonitis and admitted to the surgical unit. The intraoperative exploration found an advanced neglected peritonitis with the presence of several false membranes. A bulging appendix, inflamed and perforated, located at the pelvis in contact with the right fallopian tube which was very inflamed and swollen (1cm in diameter). An appendectomy was performed and the specimen was sent for pathological examination.
The postoperative course was simple and the patient was discharged at 6 days postoperatively. The anatomopathological examination of the specimen (Figure 1 & 2) showed a histological and immunohistochemical aspect of a well-differentiated neuroendocrine tumor of grade 2. Absence of vascular emboli or peri-nerve engrainments. The limit of excision is tumoral and the tumor is classified as pT3.
The patient underwent a thoracoabdomino-pelvic CT scan (Computed Tomography scan) which was free of any suspicious abnormalities, notably no evidence of tumor residue at the RIF or any pathological contrast on arterial time.
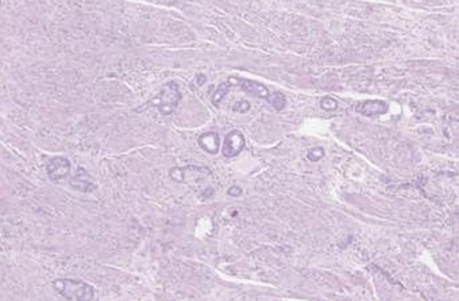
Figure 1: Histopathological images of the appendix
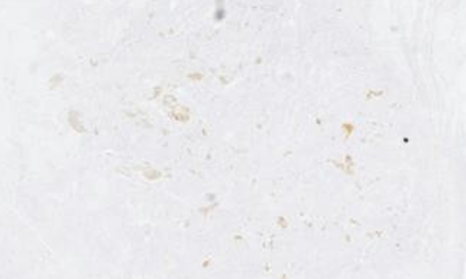
Figure 2: Immunohistochemistry of the appendix (positive for chromogranin A)
Discussion
Neuroendocrine tumors represent a heterogeneous group of tumors developed from the disseminated neuroendocrine system (DENS) with common morphological and immunohistochemical characteristics.
NETs can occur throughout the body, but are predominantly found in the gastroenteropancreatic system (73.4%) [4]. They occur mainly in the small intestine (44.7%), rectum (19.6%), appendix (16.7%), colon (10.6%) and stomach (7.2%) [5].
The annual incidence of neuroendocrine tumors of the appendix is estimated to be 0.15 up to 0.6 per 100,000 people according to the SEER (Surveillance Epidemiology and End Results) data [6]; with a slight female predominance. The peak incidence in women is between 15 and 19 years of age and between 20 and 29 years of age in men [7].
The rarity of clinical signs makes the preoperative diagnosis of neuroendocrine tumors of the appendix very difficult. They are most often discovered by chance on a surgical excision specimen (0.3 to 0.5% of specimens) [6], as was the case for our patient.
The diagnosis of certainty remains histological. A classification of NETs has been developed by the World Health Organization and updated in 2017 [8] (Figure 3). This classification takes into account the degree of cellular differentiation. Differentiated tumors have a low proliferative potential, in contrast to poorly or undifferentiated carcinomas.
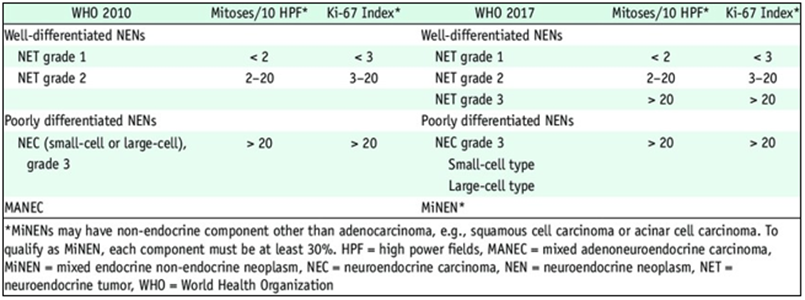
Figure 3: WHO Classification of digestive neuroendocrine tumors: Comparison to WHO 2010 and 2017NETs are characterized by the presence of chromatin clots. The particular phenotype of these cells contributes to the diagnosis during immunohistochemistry examinations. Indeed, the following markers are expressed with varying degrees of specificity: Synaptophysin, NSE, chromogranin A [9]. The presence of at least two of these markers allows the diagnosis of neuroendocrine carcinoma.
Neuroendocrine tumors of the appendix generally have a good prognosis. It accounts for 32- 57% of appendicular tumors [10]. In 70-90% of cases, they are smaller than 1 cm, with a very low risk of metastasis, whereas this risk is 30-60% when the tumor is larger than 2 cm [10]. The histological type and mesoappendix invasion can be also considered as predictive factors of aggressiveness.
The treatment is mainly surgical depending on the size of the tumor [11].
- An appendectomy with complete resection of the mesoappendix is sufficient. For a tumor smaller than 1cm.
- A right hemicolectomy with lymph node dissection is indicated after checking for liver metastases:
• For tumors larger than 2cm.
• When the tumor is located at the base of the appendix.
• When there are venous or lymphatic emboli.
• Tumors with a high grade of malignancy.
The medical treatment of metastatic tumors or those with carcinoid syndrome involves two types of medication: Chemotherapy and anti-secretory treatments, based on somatostatin analogues, which are very effective on the clinical symptoms of carcinoid syndrome, but their anti-proliferative effect remains controversial (stabilization of tumor growth in 50% of cases according to some studies) [12]. Local treatments can also be used: hepatic arterial chemoembolization for diffuse unresectable liver metastases and radiotherapy.
Conclusion
The absence of clinical signs in the majority of cases makes the early diagnosis of neuroendocrine tumors of the appendix very difficult, and almost always postoperative. Hence the importance of systematically sending any appendectomy specimen for pathological examination.
References
- Masson, E. (2022). Tumeurs endocrines digestives : stratégie diagnostique [Internet]. EM-Consulte. [cité 22 avr 2022]. Disponible sur: https://www.em-consulte.com/article/132721/tumeurs-endocrines-digestives-strategie-diagnosti
View at Publisher | View at Google Scholar - A G. (1914). Tumeurs endocrines de l’appendice. Press Med, 22, 37‑40.
View at Publisher | View at Google Scholar - Scoazec, J. Y., & Couvelard, A. (2011). pour le réseau TENpath (réseau national d’expertise pour le diagnostic anatomopathologique des tumeurs neuroendocrines malignes de l’adulte, sporadiques et familiales).[The new WHO classification of digestive neuroendocrine tumors]. Ann Pathol, 31(2), 88-92.
View at Publisher | View at Google Scholar - Modlin, I. M., & Sandor, A. (1997). An analysis of 8305 cases of carcinoid tumors. Cancer, 79(4), 813-829.
View at Publisher | View at Google Scholar - Maggard, M. A., O'Connell, J. B., & Ko, C. Y. (2004). Updated population-based review of carcinoid tumors. Annals of surgery, 240(1), 117.
View at Publisher | View at Google Scholar - Pape, U. F., Perren, A., Niederle, B., Gross, D., Gress, T., Costa, F., ... & Grossman, A. (2012). ENETS Consensus Guidelines for the management of patients with neuroendocrine neoplasms from the jejuno-ileum and the appendix including goblet cell carcinomas. Neuroendocrinology, 95(2), 135-156.
View at Publisher | View at Google Scholar - Pawa, N., Clift, A. K., Osmani, H., Drymousis, P., Cichocki, A., Flora, R., ... & Frilling, A. (2018). Surgical management of patients with neuroendocrine neoplasms of the appendix: appendectomy or more. Neuroendocrinology, 106(3), 242-251.
View at Publisher | View at Google Scholar - RV L, RY O, G K, J R. WHO Classification of Tumours of Endocrine Organs [Internet]. [cite 22 avr 2022]. Disponible sur: https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/WHO- Classification-Of-Tumours-Of-Endocrine-Organs- 2017
View at Publisher | View at Google Scholar - Kaltsas, G. A., Besser, G. M., & Grossman, A. B. (2004). The diagnosis and medical management of advanced neuroendocrine tumors. Endocrine reviews, 25(3), 458-511.
View at Publisher | View at Google Scholar - Tchana-Sato, V., Detry, O., Polus, M., Thiry, A., Detroz, B., Maweja, S., ... & Honoré, P. (2006). Carcinoid tumor of the appendix: a consecutive series from 1237 appendectomies. World journal of gastroenterology: WJG, 12(41), 6699‑6701.
View at Publisher | View at Google Scholar - Goede, A. C., Caplin, M. E., & Winslet, M. C. (2003). Carcinoid tumour of the appendix. Journal of British Surgery, 90(11), 1317-1322.
View at Publisher | View at Google Scholar - Panzuto, F., Di Fonzo, M., Iannicelli, E., Sciuto, R., Maini, C. L., Capurso, G., ... & lle De Fave, G. (2006). Long-term clinical outcome of somatostatin analogues for treatment of progressive, metastatic, well-differentiated entero-pancreatic endocrine carcinoma. Annals of Oncology, 17(3), 461-466.
View at Publisher | View at Google Scholar
