

Case Report | DOI: https://doi.org/10.31579/2835-2882/003
Case Report: Neoplastic Meningitis: A Sign of Malignancy in Pituitary Tumors?
- Chasseloup Fanny 1
- Villa Chiara 2
- Hasmik Koulakian 4
- Tissier Frédérique 5
- Gaillard Stéphan 3
- Baussart Bertrand 3
- Jouanneau Emmanuel 6
- Bertherat Jérôme 1
- Raverot Véronique 7
- Raverot Gérald 8
- Assié Guillaume 1*
1 Department of Endocrinology, Cochin Hospital, Assistance Publique Hôpitaux de Paris, France; Institut Cochin, INSERM U1016 CNRS 8104 Paris Descartes University.
2 Department of Pathological Cytology and Anatomy, Foch Hospital, Suresnes, France.
3 Department of Neurosurgery, Foch Hospital, Suresnes, France.
4 Department of Radiology, Cochin Hospital, Paris.
5 Department of Pathology, Cochin Hospital, Paris.
6 Department of Neurosurgery: Pituitary Surgery. Groupement Hospitalier Est, Hospices Civils de Lyon, Bron, F-69677; INSERM U1052, CNRS UMR5286.
*Corresponding Author: Assié Guillaume, Department of Endocrinology, Cochin Hospital, Assistance Publique Hôpitaux de Paris, France; Institut Cochin, INSERM U1016 CNRS 8104 Paris Descartes University.
Citation: Chasseloup Fanny, Villa Chiara, Hasmik Koulakian, Tissier Frédérique, Gaillard Stéphan. et al. (2022). Neoplastic Meningitis: a Sign of Malignancy in Pituitary Tumors? International Journal of Clinical Research and Studies.1(1); DOI:10.31579/ijcrs-2022/003
Copyright: © 2022 Assié Guillaume. This is an open-access artic le distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 29 September 2022 | Accepted: 07 October 2022 | Published: 17 October 2022
Keywords: neoplastic meningitis; pituitary tumor; pituitary carcinomas; cushing disease; prolactinoma
Abstract
Context: Pituitary carcinomas are rare. Distant metastases are required for positive diagnosis. Most often pituitary metastases occur within the central nervous system, suggesting dissemination through cerebrospinal fluid. However, meningitis reaction, though common in neoplastic meningitis of other origin, is not reported in pituitary tumors.
Objective: To report two cases of meningitis in the context of aggressive pituitary tumors.
Patients and methods: Two patients with neoplastic meningitis are reported. These patients presented invasive pituitary tumors, characterized by hormone assays, MRI, lumbar puncture and pathology.
Results: Patient 1 presented with an early recurrence of Cushing syndrome in the context of an atypical ACTH-secreting pituitary tumor. Meningitis appeared soon after. Lumbar puncture showed meningitis, with malignant cells confirmed by ACTH immunocytochemistry. Patient 2 presented an atypical prolactin-secreting pituitary tumor. This patient also presented meningitis, confirmed by lumbar puncture. Both patients responded poorly to standard treatments, with a fatal evolution related to the progression of their pituitary disease.
Conclusion: These two cases illustrate the occurrence of meningitis in the context of aggressive pituitary adenomas, and raise the question whether meningitis is not underdiagnosed in this setting.
Abbreviated Title: Neoplastic meningitis and Pituitary Carcinomas
Introduction
Pituitary carcinomas are defined in the 2017 World Health Organization (WHO) classification by the existence of distant metastases [1]. Pituitary carcinomas are rare, about 0.2 to 0.5 % of all pituitary tumors [2-4]. In general, these tumors are invasive to adjacent structures, and show a rapid growth, with insufficient response to standard treatment or early relapse. Dissemination may occur via lymphatic and hematogenous spreading. Cerebrospinal dissemination may also occur, as suggested by the common locations in the central nervous system, either the brain or the medulla. Systemic spread occurring via blood diffusion could be linked to invasion of the cavernous sinus by the pituitary tumor [2,5,6].
Survival with pituitary carcinoma is limited, calling for an early diagnosis for early treatment attempts. Systemic chemotherapy is discussed in such situations, mainly with temozolomide [7,8].
Neoplastic meningitis is common in the setting of malignant blood diseases, embryonal and germ cell tumors of the central nervous system and some epithelial tumors such as breast, lung and melanoma. Symptoms may be nonspecific. Clinical signs of meningitis are often mild, so diagnosis is delayed. In the setting of pituitary tumors, neoplastic meningitis is not well characterized. Only one case reported pituitary tumor cells in the CSF, more than 30 years ago [9]. Beyond this case, meningitis in the context of aggressive pituitary tumors has not been the focus of attention in medical literature with no specific reports. Here we report two cases of aggressive pituitary tumors with meningitis.
Patients and Methods
Patients’ characterization: Patients were included from two French centers (Cochin hospital in Paris, and Lyon), followed between 2008 and 2012.
Imaging: Pituitary MRI scans were performed with coronal and sagittal T1 weighting, with and without enhancement and with coronal T2 weighting with 1.5T.
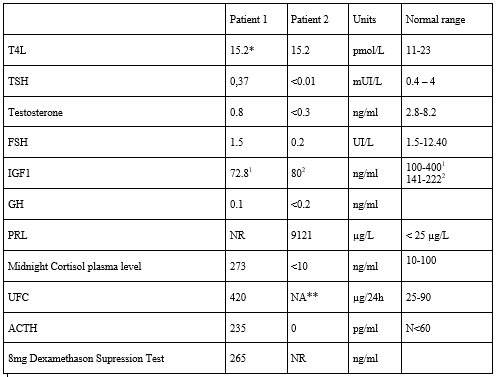
Hormonal assessment: ACTH was assayed by immunoradiometric assay (ELSA-ACTH, Cis Bio International, Gif-sur-Yvette, France) and cortisol was assayed by competition assays (IMMULITE 2000 Cortisol; Diagnostic Products Corp., Los Angeles, USA) for patient 1. Prolactin was assayed by radioimmunoassay and chemiluminescence (VIDAS Biomerieux) for patient 2.
CSF Analysis: 30 ml of CSF was sampled by lumbar puncture. Routine CSF analysis was performed, including cell counts, glucose and protein assays, then completed by pathology analysis after centrifugation. Pathology analysis of LCR was realized by May-Grünwald Giemsa staining and ACTH immunocytochemistry (Antibody Clone 02A3, Dako Cytomation, Carpinteria, CA).
Results
Patient 1
A 61-year-old man presented with rapid diminution of visual acuity and quadranopsia. Past medical history was marked by bilateral chronic glaucoma, high blood pressure, and vertebral compression fracture.
A cerebral and pituitary Magnetic Resonance Imaging (MRI) was performed, showing a 30x20 mm pituitary tumor, with right parasellar extension and invasion of the right cavernous sinus (Fig 1A). Diagnosis of Cushing syndrome was suggested by amyotrophic limbs, high blood pressure, vertebral fracture, and hypokalemia. Biological confirmation was obtained, with elevated UFC (urinary free cortisol) of 7xULN (upper limit of normal), and elevated early morning plasma ACTH (42 pmol/l; normal: 1 to 10; Figure 1B).
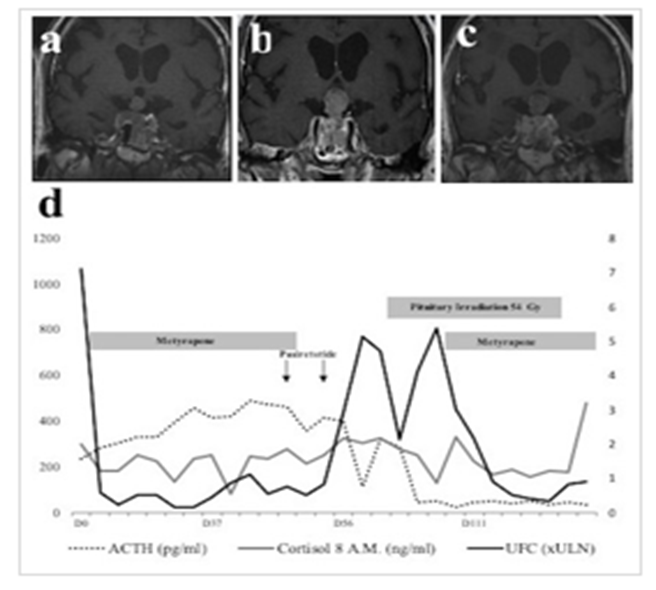
A, B and C: panel A, B and C shows MRI at day 5, day 68 and day 119 after initial diagnosis. D: panel D shows evolution of ACTH (dash grey line), 8 a.m. serum cortisol levels (full black line) and urinary free cortisol (UFC) (full black line). ACTH values are provided as pg/ml (left axis). 8 A.M serum cortisol levels are provided as ng/ml (left axis). UFC is expressed as upper limit of the normal range (ULN) on the right axis.
A first transsphenoidal pituitary surgery trial was aborted due to a left carotid artery wound. After, the patient developed a bilateral phlebitis and a pulmonary embolism. Treatment included anticoagulation along with an inferior vena cava filter.
A second transsphenoidal procedure was performed one month later. An intrasellar portion of the lesion was removed. Pathological examination revealed an ATCH-secreting adenoma, with a high degree of atypia, high proliferation index (Ki67/MIB1:30%), brisk mitotic activity (9/10HPF), and strong expression of p53 (60%). The tumor was thereafter referred to as an atypical adenoma according to the 2004 WHO classification.
After surgery, the patient presented with ACTH and TSH deficiencies. The postoperative period was complicated by Pseudomonas aeruginosa pneumonia, with septicemia and acute respiratory distress syndrome. The patient was subsequently transferred to the intensive care department, where he stayed for two months. He subsequently developed critical illness polyneuropathy.
Bone scintigraphy, CT-scan of the chest, abdomen and pelvis did not reveal any secondary localization.
Around 110 days after initial diagnosis, the patient presented signs of relapse and was admitted to the hospital.
Relapse was proven with an elevated UFC - 420 µg/24h (normal, 25-90)-, a high plasma midnight cortisol - 273 ng/ml (normal: 10 to 100 ng/ml)-, a flat cortisol cycle - with plasma cortisol at 233 ng/ml at 0800 h and 273 ng/ml at 0000 h. In addition, plasma cortisol and ACTH secretion were not suppressed 8 hours after 8mg of dexamethasone at midnight – with plasma cortisol at 265 ng/ml and ACTH at 15 ng/ml-. The patient presented central hypothyroidism properly supplemented with 25 µg of Levothyroxine per day - TSH was 0.37 mUI/l (normal) and FT4 15.2 pmol/l (11-23)-, and a central hypogonadism -with serum testosterone at 0.8 ng/ml (normal, 2.8-8.2), FSH 1.5 UI/L, LH 1.3 UI/L-. Complete hormonal workup is provided in table 2.
Pituitary MRI identified a recurrence of the pituitary tumor, measured at 24x33x15 mm, with right parasellar and suprasellar extension, and contact with the optic chiasm. The right cavernous sinus was completely invaded. A medullary MRI did not show any distant metastasis. Positron Emission Tomography/Computed Tomography (PET/CT) with 18 Fluoro-Desoxy-Glucose (18-FDG) revealed a hypermetabolic pituitary tumor with no additional distant image. Medical treatment by Metyrapone was started, with a favorable clinical outcome and biological response (figure 1). Medical treatments targeting pituitary tumor growth were also started, including high-dose Cabergoline (1mg per day) and Pasireotide (600 mg x 2/day subcutaneously).
In the first day of treatment with Pasireotide the patient presented nausea, vomiting, strong headaches and paralysis of the right sixth cranial nerve. Pasireotide and Metyrapone were stopped with amelioration of symptoms. On MRI, a necrotic-hemorrhagic transformation of the tumor was observed, with no signs of intracranial hypertension. There was no indication for neurosurgical treatment. Pasireotide was reintroduced at a smaller dose (600 mg once a day) but once again the patient presented headaches and treatment was permanently discontinued and pharmacological investigation was performed.
Subsequently, radiotherapy was performed (54 Gy in 30 sessions). Disease progression was marked by recurrence of hypercortisolism with severe amyotrophy and several falls. Metyrapone was reintroduced.
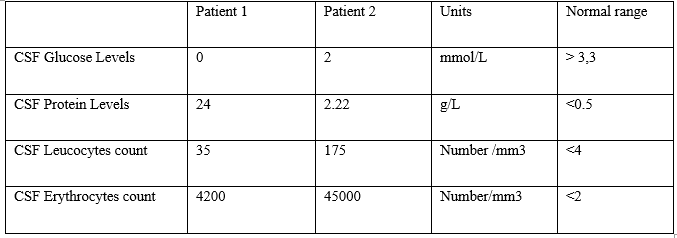
Unfortunately, disease progression was persistent, with anorexia, asthenia, confusion, and several episodes of delirium. Biological workup showed inflammatory markers, and a CT-scan showed mild hydrocephalus. A lumbar puncture was performed, both to rule out an infection, and to drain CSF (30mL), following the neurosurgeon’s advice. The CSF protein level was high at 24 g/l, glucose was low at 0 mmol/l, leucocytes increased at 35/mm3 and erythrocytes at 4200/mm3 with no bacteria on direct examination or after culture.
Pathological examination of the CSF confirmed the diagnosis of neoplastic meningitis by showing several malignant cells expressing synaptophysine, CD3-, CD20-, AE1-AE3- and PS100. ACTH immunocytochemistry was positive, with a specific cytoplasmic staining. Chemotherapy with Temozolomide was considered, but finally not started, owing to the rapid and major decline of the patient’s condition. Palliative support was administered. The patient died ten days later.
Patient 2
A 70-year-old man, with a past history of pituitary macroprolactinoma, presented with visual defects. He was diagnosed at 52 years-old with an invasive macroadenoma. His optic chiasma was compressed, inducing visual field defects. He was initially treated by dopamine agonists. However, in the absence of visual improvement, and given the limited reduction of tumor burden, pituitary surgery was performed 6 months later. Pathological examination reported a lactotroph adenoma with invasion of the respiratory mucosa, low Ki67 proliferation index, low p53 expression and 5 mitosis/10HPF. Surgery was followed by conventional radiotherapy. The following years were marked by resistance to dopamine agonists including Carbergoline, Bromocriptine and Quinagolide, despite high doses. In this context, a second conventional radiotherapy was performed fifteen years later (38 Gy). Despite this treatment, two years later, the patient presented with diplopia and peri-orbital neuralgia. On MRI, the tumor had grown, invading both cavernous sinuses (figure 2). At this time, no metastasis was found on whole-body CT-scan, 18-FDG PET/CT and medullary MRI.Somatostatin analogues were started but rapidly stopped due to side effects. Temozolomide (300mg per day for five days, every 28 days) was then introduced. Diplopia and neuralgia improved. Prolactin and tumor volume decreased (figure 2) after 3 cycles.
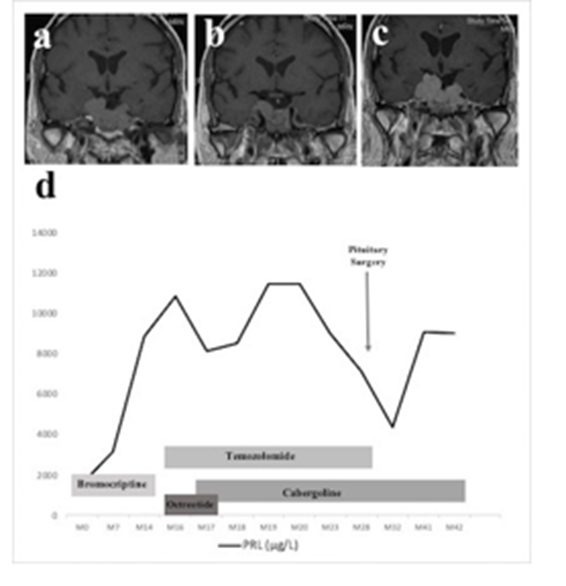
A, B and C: panel A, B and C shows MRI at 7, 17 and 32 months after recurrence. D: panel D shows evolution of prolactin (full black line). Prolactin values are provided as ug/L (left axis). Despite initial efficacy, prolactin progressively increased (figure 3). Thrombocytopenia appeared and Temozolomide was stopped after eight cycles. Rapidly thereafter, diplopia recurred. An MRI showed the tumor had increased (figure 3). A new surgery was performed. The pathological report showed an increased Ki67 proliferation index (11%) compared to the first surgery, p53 expression but mitotic activity was lower than 1/10 HPF. Diplopia improved and prolactin levels decreased.
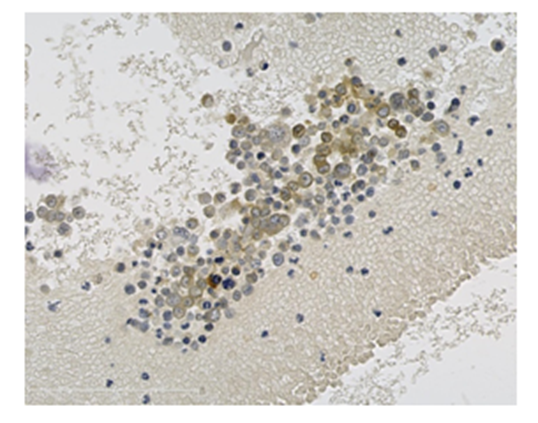
After centrifugation, CSF cells were stained with anti-ACTH immunocytochemestry. ACTH positive cells are shown in brown.
A few months later, the patient presented with an episode of brutal headache and vomiting. The diagnosis of meningitis was evoked, and a lumbar puncture was performed. CSF analysis showed 175 elements, with cellular polymorphism, increased proteins at 2.22 g/L, and low glucose level at 2.0 mmol/L (table 1). No bacteria were found, neither on direct examination nor after culture. No tumor cell was observed.
After lumbar puncture, symptoms improved, but intracranial hypertension worsened. Evolution was rapidly progressive, and the patient died in the presence of altered alertness and inhalation pneumonia. Autopsy was performed. The sellar region was diffusely invaded by the adenoma; in particular the sellar roof, sphenoidal and cavernous sinus. No evidence of central nervous system or visceral metastasis was reported.
Discussion
These cases illustrate meningitis reactions in the situation of aggressive pituitary tumors. These two patients were not diagnosed with distant metastases. And according to the standard 2017 WHO classification, distant metastases are required to ascertain malignancy. In these two cases, shouldn’t we consider the pituitary tumors as malignant? Especially in one case we could identify neoplastic cells in CSF. In terms of CSF biology, neoplastic meningitis is typically associated with increased leukocytes (>4/mm3), elevated protein (>0.5 g/L) and decreased glucose (<3>
Neoplastic meningitis can be diagnosed by MRI. Radiological signs of meningitis include contrast enhancement and thickening of meninges, resulting in a fine intense signal layer 10. In our patients, no sign of meningitis was observed on MRI. This is probably related to the limited sensitivity of MRI for diagnosing neoplastic meningitis. Indeed, MRI is normal in 30% of cases of neoplastic meningitis [11].
Pathogenesis of pituitary carcinomas is still unclear. Malignant cells potentially spread either via craniospinal fluid, lymphatic or by haematogenous routes 3. Since CNS localizations are the most common metastatic sites in pituitary carcinomas, one may speculate that CSF is a privileged path for dissemination of tumor cells. Though there is no demonstration of systematic meningitis reaction when tumor cells spread to CSF, it is reasonable to consider that this meningitis reaction occurs at least in some cases. If we consider the potentially limited manifestations of neoplastic meningitis and the similarity observed between neoplastic meningitis signs and atypical pituitary tumors symptoms, shouldn’t lumbar puncture be performed more frequently when new neurological signs occur during atypical pituitary tumor follow up?
In clinical oncology, when neurological signs occur, physicians are usually aware of the possibility of neoplastic meningitis. Neoplastic meningitis can manifest in different ways: hemisphere dysfunction (headache, mental status or cognitive impairment), cranial nerve dysfunction (diplopia, trigeminal sensory or motor loss, or optic neuropathy) and spinal cord signs (neck pain, limb weakness or radicular sensory loss). However nuchal rigidity is present in only 15 % of cases [10,11]. The first patient presented here had hemisphere dysfunction and spinal cord signs with confusion, delirium and motor deceleration. The second had cranial nerve dysfunction with diplopia and neuralgia, not considered as related to a cavernous syndrome. Nuchal rigidity was absent in both cases. Symptoms of neoplastic meningitis and pituitary tumors can overlap, leading to undiagnosed neoplastic meningitis in the context of atypical pituitary tumors.
In the literature, meningitis is not reported in pituitary carcinomas. Instead, previous publications focused on the description of distant metastases and on the features of the primary tumor. Distant metastases in pituitary tumors have been reported after a mean period of 8 years following the initial diagnosis of pituitary tumor [3,6,12]. These metastases occurred mainly in the central nervous system (45% of cases), but also out of the CNS (39% of cases), or both (16,1%) [3,13]. Beyond the metastasis, features of the pituitary carcinomas are not specific. Indeed, in terms of local behavior, local invasion is commonly observed in adenomas -95% of macroadenomas and 66 % of microadenomas for dura mater- and is not systematically associated with aggressiveness and resistance to treatments [6,13,14]. Efforts have been made to search for histological markers discriminating atypical adenomas from carcinomas. Ki67 immunohistochemistry, reflecting tumor proliferation index, has a limited prognostic value [15,16]. Another potential marker is p53 immunohistochemistry but it is still controversial. Discordant studies have been reported about the prognostic value of this marker. However, a grading system based on invasion on magnetic resonance imaging, immunocytochemical profile, Ki-67, mitotic index, and p53 positivity, allows a better classification of pituitary tumor patients who have a high risk of early recurrence or progression [17]. Despite this improvement, pathological markers are unable to identify pituitary carcinomas. Indeed, in pituitary carcinomas -documented with metastasis-, p53 expression is highly variable, ranging from 0 to 30 % [18]. In one study, p53 is higher in metastases (88%) than in primary (57%) [6]. A recent study has compared carcinomas and atypical adenomas. A 2% p53 positivity seems the best threshold to discriminate atypical adenomas from typical adenomas. However, p53 cannot discriminate carcinomas from atypical adenomas [19].
The two cases presented here illustrate the possible occurrence of neoplastic meningitis in the context of atypical pituitary tumors. If present, neoplastic meningitis could be considered a sign of malignancy, and thus could change the course of treatment.
References
- Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol (Berl). 2017;134(4):521-535.
View at Publisher | View at Google Scholar - Kaltsas GA, Nomikos P, Kontogeorgos G, Buchfelder M, Grossman AB. Diagnosis and Management of Pituitary Carcinomas. J Clin Endocrinol Metab. 2005;90(5):3089-3099.
View at Publisher | View at Google Scholar - Lopes MBS, Scheithauer BW, Schiff D. Pituitary carcinoma. Endocrine. 2005;28(1):115-121.
View at Publisher | View at Google Scholar - Heaney AP. Pituitary Carcinoma: Difficult Diagnosis and Treatment. J Clin Endocrinol Metab. 2011;96(12):3649-3660.
View at Publisher | View at Google Scholar - Tysome J, Gnanalingham KK, Chopra I, Mendoza N. Intradural metastatic spinal cord compression from ACTH-secreting pituitary carcinoma. Acta Neurochir (Wien). 2004;146(11):1251-1254.
View at Publisher | View at Google Scholar - Pernicone PJ, Scheithauer BW, Sebo TJ, et al. Pituitary carcinoma. Cancer. 1997;79(4):804-812.
View at Publisher | View at Google Scholar - Hansen TM, Batra S, Lim M, et al. Invasive adenoma and pituitary carcinoma: a SEER database analysis. Neurosurg Rev. 2014;37(2):279-286
View at Publisher | View at Google Scholar - Raverot G, Sturm N, de Fraipont F, et al. Temozolomide Treatment in Aggressive Pituitary Tumors and Pituitary Carcinomas: A French Multicenter Experience. J Clin Endocrinol Metab. 2010;95(10):4592-4599.
View at Publisher | View at Google Scholar - Hashimoto N, Handa H, Nishi S. Intracranial and intraspinal dissemination from a growth hormone-secreting pituitary tumor: case report. J Neurosurg. 1986;64(1):140-144.
View at Publisher | View at Google Scholar - Chamberlain MC. Neoplastic Meningitis. The Oncologist. 2008;13(9):967-977.
View at Publisher | View at Google Scholar - Hyun J-W, Jeong IH, Joung A, Cho HJ, Kim S-H, Kim HJ. Leptomeningeal metastasis: Clinical experience of 519 cases. Eur J Cancer. 2016;56:107-114.
View at Publisher | View at Google Scholar - van der Klaauw AA, Kienitz T, Strasburger CJ, Smit JWA, Romijn JA. Malignant pituitary corticotroph adenomas: report of two cases and a comprehensive review of the literature. Pituitary. 2009;12(1):57-69.
View at Publisher | View at Google Scholar - Kaltsas GA, Grossman AB. Malignant pituitary tumours. Pituitary. 1998;1(1):69-81.
View at Publisher | View at Google Scholar - William A.S. Taylor, F.R.C.S.(S.N.), David Uttley, F.R.C.S., and P.R.Wilkins, M.R.C.Path. Multiple dural metastases from a pituitary adenoma. Case report. J Neurosurg 81:624-626, 1994
View at Publisher | View at Google Scholar - Del Basso De Caro M, Solari D, Pagliuca F, et al. Atypical pituitary adenomas: clinical characteristics and role of ki-67 and p53 in prognostic and therapeutic evaluation. A series of 50 patients. Neurosurg Rev. May 2016.
View at Publisher | View at Google Scholar - Salehi F, Agur A, Scheithauer BW, Kovacs K, Lloyd RV, Cusimano M. KI-67 IN PITUITARY NEOPLASMS: A REVIEW-PART I. Neurosurgery. 2009;65(3):429-437.
View at Publisher | View at Google Scholar - Raverot G, Dantony E, Beauvy J, et al. Risk of Recurrence in Pituitary Neuroendocrine Tumors: A Prospective Study Using a Five-Tiered Classification. J Clin Endocrinol Metab. 2017;102(9):3368-3374.
View at Publisher | View at Google Scholar - Roncaroli F, Scheithauer BW, Young WF, et al. Silent corticotroph carcinoma of the adenohypophysis: a report of five cases. Am J Surg Pathol. 2003;27(4):477-486.
View at Publisher | View at Google Scholar - Miermeister CP, Petersenn S, Buchfelder M, et al. Histological criteria for atypical pituitary adenomas – data from the German pituitary adenoma registry suggests modifications. Acta Neuropathol Commun. 2015;3(1).
View at Publisher | View at Google Scholar
