

Case Report | DOI: https://doi.org/10.31579/2835-785X/061
Juvenile Idiopathic Arthritis in Kabuki Syndrome: A Case Report and Reviews
1 Tehran University of Medical Sciences: Tehran, Tehran, Iran.
2 Assistant Professor of Pediatric, Department of Pediatrics, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
*Corresponding Author: Fatemeh Tahghighi Sharabian, Assistant Professor of Pediatric, Department of Pediatrics, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
Citation: Atousa Dadashi, Anita Dadashi, and Fatemeh T. Sharabian (2024), Juvenile Idiopathic Arthritis in Kabuki syndrome: A Case Report and Reviews, International Journal of Clinical Research and Reports. 3(3); DOI:10.31579/2835-785X/061
Copyright: © 2024, Fatemeh Tahghighi Sharabian. This is an open-access artic le distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 04 April 2024 | Accepted: 22 April 2024 | Published: 03 May 2024
Keywords: Kabuki syndrome (KS), Juvenile idiopathic arthritis (JIA), Autoimmune disease, Characteristic face, KMT2D
Abstract
Background: Juvenile idiopathic arthritis (JIA) is a common rheumatological disorder that occurs before 16 years old. Several studies have documented an association between JIA and genetics. Kabuki syndrome (KS) is a genetic disorder that has mutations in lysine methyltransferase 2D (KMT2D) and 6A (KDM6A). However, the relationship between KS and autoimmune disease has been reported in other publications; therefore, in this paper, we report a KS case that refers to our department with arthritis.
Case report: A 6-year-old female was referred to our department with flexion contracture, swelling, and tenderness in the knees, ankles, elbows, and small joints. She had a syndromic face and mental retarded and bilateral deafness so we requested genetic test. The genetic test suggested she has a mutation in the KMT2D gene, and she was finally diagnosed with KS with JIA. She was treated with JIA to reduce symptoms and increase her quality of life.
Conclusion: KS was seen with multiple organ diseases and disabilities. We have seen an increase in reports of autoimmune disease in KS. A hypothesis in this paper is that JIA and other autoimmune diseases are associated with KMD2T mutations.
Introduction
Kabuki syndrome (KS) is a genetic and clinical syndrome that was first described in 1981 in Japanese by Niikawa and Kuroki [1]. Kabuki syndrome was characterized by five manifestations, which included: facial appearance (including eversion of the lower lateral eyelid, a depressed nasal tip, arched eyebrows with the lateral one third dispersed, and prominent ears). postnatal short stature, skeletal anomalies, moderate mental retardation, and dermatoglyphic anomalies (2]. Kabuki syndrome susceptible patients to autoimmune and infection disease. Autoimmune manifestations that occur with KS include idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, autoimmune thyroiditis, vitiligo, DM, celiac, and Chron's disease [3].
Case Presentation
A 6-year-old female was referred to a children’s hospital in Tehran, Iran, on October 20, 2022. She is a single child in a family and was born by cesarean section due to a decelerating fetal heart rate. She was born term, and her birth weight was 3200 grams. She was a healthy mother without any medical history of pregnancy, and she had a healthy father. She has flexion contractures, edema, swelling, tenderness in her ankles, elbows, PIP joints, knees, fever, and a development delay. She has facial dysmorphism, an undiagnosed syndrome, a history of bilateral deafness, and poor feeding during the infancy period. On physical examination: depressed nasal tip, arched eyebrows, long eyelashes, long palpebral fissure with eversion of the lower eyelid, micrognathia (Shown in figure 1), mental retardation, and postnatal growth deficiency, Examination of joints shows full flexion in knees and elbows, swelling, and tenderness. Other examinations, such as heart, lung, abdomen, etc., were normal and did not show abnormalities.
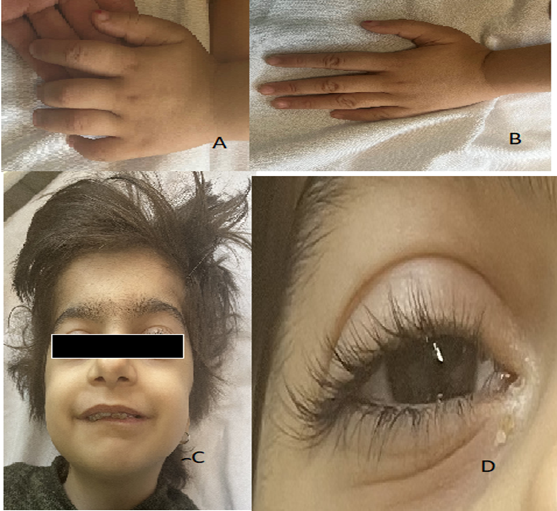
Figure 1: (A) flexion in small joints, before treatment, (B)(extension in small joint, after treatment), (C) characteristic face, (D) long eyelashes
Laboratory findings included elevated ESR, positive FANA, negative RF, normal immunoglobulin (IgM, igG, igE), normal C3, C4, C50, anti-CCP negative, brucella test (Wrigh, Coombs wright, 2ME) negative, and other normal lab tests shown in the table.
Radiology findings included normal radiography (X-Rey) from knees, elbows, ankles, and feet and 99m Tc-MDP skeletal scintigraphy on October 20, 2022. Reported blood pool imaging revealed moderately increased activity in ankles, knees, and small foot joints. Delay images revealed fibrotic deformity of the knees with increased uptake in the right elbow, knees, ankles, and small foot joints. According to a phase-positive scan, knees, ankles, and small joints may suggest active arthropathies.
Although before referred to our hospital, according to the response to sound audiometry done and the result of this test, the last intensity that wave V observed was 90 db, bilateral severe to profound hearing loss. According to characteristic face and finding suspicion for KS, we requested a genetic test to confirm my diagnosis.
Genetic test findings
The genetic test shown KMT2D (NM:003482:exon44:c.C14036T:p.A4679V, CADD:29.9)
Treatment
Based on genetic tests and other findings, the patient was diagnosed with KS and JIA; therefore, we started treatment with methylprednisolone at a dose of 300 mg. This treatment lasted 3 days, and we continued treatment with oral prednisolone at 5mg daily, ampule Trexoma injection every ten days, and folic acid at 5mg daily and Ca daily.
Outcome and follow-up
In our patient, we saw better extension contracture and were able to sit and stand without swelling in the joints or tenderness. Our patient had a better condition during one year than on the first visit.
Discussion
We reported a case of a 6-year-old female with KS and JIA, the severe symptom of JIA in KS. The patient had a fever of unknown origin, arthritis, arthralgia, flexion contracture, and an inability to use joints, so she is not able to sit, stand, or walk. KS is a rare multisystem syndrome caused by mutations in lysine methylase 2D (KMT2D), the majority type of KS cases, about D_v, and mutations in lysine methylase 6A (KDM6A), the least type of KS case, about %1_%6, according to published KS reports [2, 4, 5]. KMT2D or MLL2 belong to a family of H3K4 methylase and demethylation of H3K4 due to disturbance of B and T cells [6, 7].
KS was first reported in 1981 in Japan by Niikawa and Kuroki. The prevalence of KS in Japan is one in every 32,000 births, with a different rate of prevalence around the world. KS is the X-linked mutation [1]. However, kabuki syndrome is a challenge because it is early diagnosed by facial characteristics such as eversion of the lower lateral eyelid, a depressed nasal tip, arched eyebrows with the lateral one-third dispersed, prominent ears [8-10], and a few kabuki cases that do not have facial characteristics [10]. Another symptom of KS is shown in older age [11].
Juvenile idiopathic arthritis is an autoimmune disease that occurs before 16 years old [12, 13]. JIA prevalence is 16–150/10000 and is not a single disease [14]. The definition of this disease is arthritis with an unknown etiology and presenting in more than one joint for more than 6 weeks [14]. JIA is a genetic disease that has been linked to cytokines [3, 15]. In autoimmune disease, we need to suppress the immune system in patients by suppressing inflammatory drugs [3, 15]. The ability to infect is reduced by immune suppression, which increases the rate of infection in these patients.
In the older published reports of autoimmune disorders in KS, such as idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, autoimmune thyroiditis, vitiligo, DM, arthritis, celiac, and Chron's disease [16, 17], In one case of JIA reported in Kabuki syndrome, we did not have evidence about the symptom, lab test, treatment, or outcome and did not discuss this case [17]. We treated our case according to the guidelines, and we reached satisfactory treatment of JIA in the KS case.
Autoimmune disease expressed in Kabuki syndrome type 1 (KMD2T) [18] that has a mutation in lysine methylase 2D has affected B-cells and T-cells, which may ultimately affect autoimmune disease. Considering the increasing incidence of autoimmune disease in KS and the onset of autoimmune disease in older age, we most frequently follow up on KS cases, and reporting the diagnosis, treatment, and outcome may help the management of KS patients to improve their quality of life. Autoimmune features present with a low level of immunoglobulin, previously associated with KS, were also normal in our case.
Previously, a study described a disorder of B-cell and T-cell differentiation in KS, our patient, and mutation gene KMT2D (type 1 Kabuki syndrome) tendency infection in patients, but in our case, we did not have a respiratory infection but a recurrent infection.
Conclusion
As we have found in this study, autoimmune disease in kabuki syndrome incidence in older ages. According to previous publications, autoimmune diseases are probably more common in Kabuki syndrome type 1.
References
- Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, et al. (1988), Kabuki make‐up (Niikawa‐Kuroki) syndrome: A study of 62 patients. American journal of medical genetics. 1988;31(3):565-589.
View at Publisher | View at Google Scholar - So PL, Luk HM, Yu KP, Cheng SS, Hau EW, Ho SK, et al. (2021), Clinical and molecular characterization study of Chinese Kabuki syndrome in Hong Kong. American Journal of Medical Genetics Part A. 2021;185(3):675-686.
View at Publisher | View at Google Scholar - Martini A, Lovell DJ, Albani S, Brunner HI, Hyrich KL, Thompson SD, Ruperto N. (2022), Juvenile idiopathic arthritis. Nature Reviews Disease Primers. 2022;8(1):5.
View at Publisher | View at Google Scholar - AWAD A. (2016), Identification of novel mutations in ASPM and KMT2D Genes responsible for Primary Microcephaly and Kabuki-like syndrome in Palestinian Families: Bethlehem University &Palestinian Polytechnic University; 2016.
View at Publisher | View at Google Scholar - Murakami H, Tsurusaki Y, Enomoto K, Kuroda Y, Yokoi T, Furuya N, et al. (2020), Update of the genotype and phenotype of KMT2D and KDM6A by genetic screening of 100 patients with clinically suspected Kabuki syndrome. American Journal of Medical Genetics Part A. 2020;182(10):2333-2344.
View at Publisher | View at Google Scholar - Goyama S, Kitamura T. (2017), Epigenetics in normal and malignant hematopoiesis: An overview and update 2017. Cancer science. 2017;108(4):553-562.
View at Publisher | View at Google Scholar - Park K, Kim J-A, Kim J. (2020), Transcriptional regulation by the KMT2 histone H3K4 methyltransferases. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 2020;1863(7):194545.
View at Publisher | View at Google Scholar - Dugan S. (2021), Kabuki syndrome. Cassidy and Allanson's management of genetic syndromes. 2021:529-538.
View at Publisher | View at Google Scholar - Shah SS, Fulton A, Jabroun M, Brightman D, Simpson BN, Bodamer OA. (2023), Insights into the genotype–phenotype relationship of ocular manifestations in Kabuki syndrome. American Journal of Medical Genetics Part A. 2023;191(5):1325-1338.
View at Publisher | View at Google Scholar - Park SJ, Ahn MB, Jang W, Cho WK, Chae HJ, Kim MS, Suh BK. (2017), A Case of Kabuki Syndrome Confirmed by Genetic Analysis: A Novel Frameshift Mutation in the KMT2D Gene. Journal of The Korean Society of Inherited Metabolic disease. 2017;17(3):103-108.
View at Publisher | View at Google Scholar - Boniel S, Szymańska K, Śmigiel R, Szczałuba K. (2021), Kabuki syndrome—clinical review with molecular aspects. Genes. 2021;12(4):468.
View at Publisher | View at Google Scholar - Rigante D, Bosco A, Esposito S. (2015), The etiology of juvenile idiopathic arthritis. Clinical reviews in allergy & immunology. 2015;49:253-261.
View at Publisher | View at Google Scholar - Barut K, Adrovic A, Şahin S, Kasapçopur Ö. Juvenile idiopathic arthritis. Balkan medical journal. 2017;34(2):90-101.
View at Publisher | View at Google Scholar - Angelis A, Tordrup D, Kanavos P. Socio-economic burden of rare diseases: a systematic review of cost of illness evidence. Health Policy. 2015;119(7):964-79.
View at Publisher | View at Google Scholar - Vastert SJ, Kuis W, Grom AA. Systemic JIA: new developments in the understanding of the pathophysiology and therapy. Best Practice & Research Clinical Rheumatology. 2009;23(5):655-64.
View at Publisher | View at Google Scholar - Arsov T, Sestan M, Cekada N, Frkovic M, Andrews D, He Y, et al. Systemic lupus erythematosus: A new autoimmune disorder in Kabuki syndrome. European Journal of Medical Genetics. 2019;62(6):103538.
View at Publisher | View at Google Scholar - Margot H, Boursier G, Duflos C, Sanchez E, Amiel J, Andrau J-C, et al. Immunopathological manifestations in Kabuki syndrome: a registry study of 177 individuals. Genetics in Medicine. 2020;22(1):181-8.
View at Publisher | View at Google Scholar - Leonardi L, Testa A, Feleppa M, Paparella R, Conti F, Marzollo A, et al. Immune dysregulation in Kabuki syndrome: a case report of Evans syndrome and hypogammaglobulinemia. Frontiers in Pediatrics. 2023;11:1087002.
View at Publisher | View at Google Scholar
