

Review Article | DOI: https://doi.org/10.31579/2835-8376/042
Beyond Leptin and Adiponectin: The Diverse Roles of Adipokines in The Myocardial Hypertrophic Process and Heart Failure and Their Potential Contribution in Obesity
- Morris Karmazyn *
- Xiaohong Tracey Gan
Department of Pharmacology and Physiology, University of Western Ontario, London, Ontario, Canada N6A 5C1.
*Corresponding Author: Morris Karmazyn, Department of Pharmacology and Physiology, University of Western Ontario, London, Ontario, Canada N6A 5C1
Citation: Morris Karmazyn, Xiaohong Tracey Gan., (2025). Title Beyond Leptin and Adiponectin: The Diverse Roles of Adipokines in the Myocardial Hypertrophic Process and Heart Failure and their Potential Contribution in Obesity, Clinical Research and Reviews.4(2); DOI: 10.31579/2835-8376/042.
Copyright: © 2025, Morris Karmazyn, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 11 March 2025 | Accepted: 26 March 2025 | Published: 16 April 2025
Keywords: adipokines; adipocytokines; obesity; cellular signalling; cardiac hypertrophy and remodelling; heart failure
Abstract
It is now widely recognized that adipocytes have the ability to produce a myriad of bioactive compounds released into the circulation and affecting distal organs including the heart. These factors, termed adipokines, are also produced by various tissues in addition to adipocytes including cardiac tissue and have the ability to modulate cardiac function and the response to pathology. Among the processes greatly affected by adipokines is myocardial remodelling due to hypertrophy and fibrosis, two processes which contribute to the development of heart failure. This is particularly relevant under conditions of obesity and the accompanied increased adiposity in general resulting in increased adipokine production. The effects of adipokines on cardiac remodelling can be both beneficial or adverse, depending on adipokine type such as adiponectin and leptin, respectively. The molecular bases underlying the effects of adipokines on myocardial remodelling have been extensively studied and likely involved a multiplicity of cell signalling processes thus demonstrating substantial complexity. Emerging evidence suggests that these proteins play an important role in cardiac pathology. Their precise contribution is yet to be determined with certainly as this likely reflects a balance between pro-remodelling and anti-remodelling factors.
Introduction
It is now widely recognized that adipocytes function as endocrine organs and possess the capability to produce and release into the circulation a plethora of bioactive proteins that affect numerous aspects of human biology and homeostasis related to both physiology and pathophysiology. These proteins, termed adipokines demonstrate an almost bewildering array of functions influencing numerous organ system. More than 100 adipokines have thus far been identified among which leptin and adiponectin arguably representing the most widely known of these factors. Adipokines, once released into the circulation, have the ability to modulate various disease processes by acting on specific receptors in distal organs in an endocrine manner which results in alterations in cellular signalling. This is clearly the case with respect to the heart which serves as a target for the actions of many adipokines, and indeed can synthesize a variety of adipokines. These cardiac-derived adipokines can then then function as well in an autocrine or paracrine manner to alter cardiac function. Nonetheless, adipocytes serve as the primary source of adipokines and, accordingly, the production of adipokines is greatly affected by the degree of adiposity with generally increased levels seen in obese individuals. Obesity is a major risk factor for the development of heart disease in general including heart failure which occurs as a result of myocardial remodelling due to cellular hypertrophy and increased fibrosis. The question arises as to whether adipokines participate in either a negative or positive manner in the development of heart failure especially in obese individuals where their production, at least with respect to most adipokines, is elevated. Is the increased risk of heart failure in obese subjects due to the production of adipokines? As discussed here, extensive evidence implicates adipokines in the myocardial remodelling process either by promoting myocardial remodelling or by functioning in a protective manner. It is widely believed that the balance between levels of pro- and antihypertrophic adipokines will determine their net effect on myocardial remodelling and the eventual development of heart failure. A comprehensive understanding of the complexity of adipokines in terms of their mechanisms of action is rendered difficult by the sheer number of adipokines thus far identified and by the fact that each individual adipokine appears to affect multiple cell signalling and molecular events. Unravelling the key cellular mechanisms involved in the cardiac actions of adipokines represents a major challenge. In view of the emerging convincing evidence implicating adipokines in the heart failure process we focus here on a number of major adipokines and their potential roles in the development of heart failure particularly, although not exclusively, as this relates to myocardial remodelling and heart failure associated with obesity and how this knowledge can be applied to therapeutic intervention.
Obesity And Heart Disease with Particular Emphasis on Myocardial :
Remodeling And Heart Failure:
The role of obesity as a major risk factor for many diseases has been recognized for many years [reviewed in 1,2]. Obese individuals, officially defined as those with a BMI of 30 kg/m2 or greater, is considered a disease by the WHO. According to WHO data, 1 in 8 people in the world was living with obesity in 2022 and worldwide adult obesity rates have more than doubled since 1990 (https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight). One of the major consequences among various pathologies at increased risk in obese individuals is heart and other cardiovascular conditions which encompass the development of heart failure [3,4]. While it is beyond the scope of this review to discuss the primary mechanisms which account for increased cardiovascular risk accompanying obesity in general, a number of well-established factors can be noted, as illustrated in Figure 1, including dyslipidemia, increased incidence of Type 2 diabetes as well as hypertension [5]. Many factors dictate the severity of cardiovascular risk under obese conditions among these being the localization of the increased adiposity. For example, increased pericardial and visceral abdominal fat deposition has been identified as particularly important risk factors for the development of cardiovascular disease including heart failure [6,7]. Indeed, epicardial adipose tissue deposition has been identified specifically as a critical factor in the development of heart failure with HFpEF [8,9].
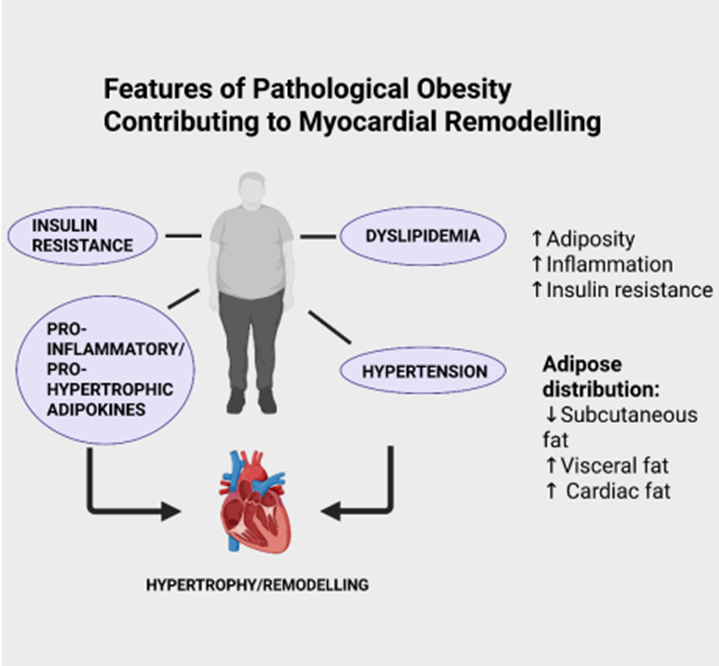
Figure 1: Some factors which contribute to myocardial remodelling and heart failure under conditions of obesity. Created with BioRender.com. As is evident for cardiovascular disease in general, many factors have been implicated in the increased incidence of heart failure under conditions of obesity including cardiac dysfunction and structural abnormalities, increased arrhythmogenesis, hypertension, cardiac lipotoxicity, insulin resistance, vascular endothelial injury and others [10,11]. The release of biologically active factors released from adipose tissue has also been proposed as potentially regulating myocardial remodelling although, as will be discussed in this review, in either deleterious or beneficial manner. These diverse effects of adipokines reflect the plethora of compounds produced by adipocytes each with distinct biological properties. Moreover, the nature of the fat deposition, e.g. visceral vs subcutaneous distribution could dictate the overall influence of obesity on the cardiovascular system particularly as visceral fat is known to produce adipokines of the more deleterious phenotype with lesser amounts of protective adipokines when compared to subcutaneous fat deposits [12,13]. Thus, the nature of body fat distribution is a critical factor when addressing the question of underlying mechanisms contributing to increased cardiovascular risk with obesity. 3. The ”Obesity Paradox”: a Role for Adipokines? Although obesity is generally considered as an important contributor to heart disease including heart failure, the concept of the obesity paradox has been proposed by numerous authors and dictates that in a sub group of patients, obesity may be salutary and lead to a reduction in mortality in patients with heart failure [14–16].The mechanisms underlying the obesity paradox are unclear but may be related to the nature and location of the adiposity which could favour production of beneficial subcutaneous adipocyte-derived adipokines resulting in cardioprotection [17,18]. Indeed, as will be discussed here, a number of adipokines including adiponectin and others, have been shown to have antiremodelling and antihypertrophic effects and therefore these could represent important contributors to the obesity paradox phenomenon. While clearly representing an interesting and potentially important phenomenon helping to more fully understand the complex role of obesity in heart disease, it should be noted that others have questioned the concept of the obesity paradox and have shown that this proposed phenomenon is non existent when assessing obesity in subjects using newer anthropometric measurements of obesity rather than BMI, such as waist-to-height ratio, relative fat mass, and body roundness index and, additionally, when correcting for other prognostic variables [19]. Indeed, when applying these criteria to more than 1000 patients with heart failure with reduced ejection fraction, obesity was associated with increased mortality in these individuals [19]. Thus, the obesity paradox remains an overall conceptually interesting phenomenon that requires substantial future investigations to determine its validity and specifically, the potential contribution, if any, of “cardiac friendly” adipokines.
Adipocytes As Endocrine Organs:
The concept of adipose tissue as a source of biologically active factors was first proposed in 1948 [20] and subsequently reviewed by a number of authors [e.g. 21-23]. Although leptin, first identified in 1994, holds an important distinction as the most studied adipose-derived factor to be identified with nearly 47000 publications, likely due to its satiety influence, and one which exerts diverse and extensive biological effects, it is recognized today that adipocytes have the ability to produce and secrete a plethora of biologically active proteins generally referred to as adipokines or also adipocytokines. This knowledge has led to the genesis of a relatively novel and highly active area of biological research termed adipobiology which has been proposed as a potential target for therapeutic intervention [24]. The science of adipobiology is based on the concept that adipocytes, particularly WATs, are not simply structural units involved in fat storage but rather, as originally proposed by Wertheimer and Shapiro, also important modulators of energy metabolism [20]. Since the discovery of a myriad of adipokines it is now widely accepted that adipocytes are in effect endocrine organs capable of modulating an extensive array of physiological functions through the release into the circulation of specific adipokines. It is interesting to point out that although leptin represents the most studied of the adipokines it is not the first of such compounds to be identified, an honour which belongs to adipsin, discovered in 1987 (see below). As we shall note, these diverse effects of adipokines may represent both salutary as well as deleterious influences on biological properties, particularly with regards to the cardiac hypertrophic program. Although WATs are considered as the primary source of adipokines, BATs, considered primarily as regulators of thermogenesis, can also produce and release bioactive proteins into the circulation such as neuregulin 4 (discussed below), known as batokines [25]. A concept important to underscore is that many adipokines, although considered primarily as adipocyte-derived actors, can also be produced by cells of non-adipocyte tissues. This is particularly the case with respect to the cardiovascular system where adipokines have been produced and released by the heart as well as vasculature, as discussed below. However, as adipokines are principally derived from adipocytes, this has led to extensive research into their role, either beneficial or deleterious, in cardiac pathology, as well as pathologies of other organ systems in obesity as the latter is obviously associated with increased adiposity and resultant dyslipidemia which contributes to cardiovascular disease [26]. Due to the large number of adipokines thus far identified a general role of adipokines in pathology is difficult to assign with precision particularly in view of the myriad of effects that these proteins exert. As will be discussed, this quandary certainly applies to their contribution to myocardial hypertrophy and remodeling.
Obesity, Myocardial Remodeling and Heart Failure :
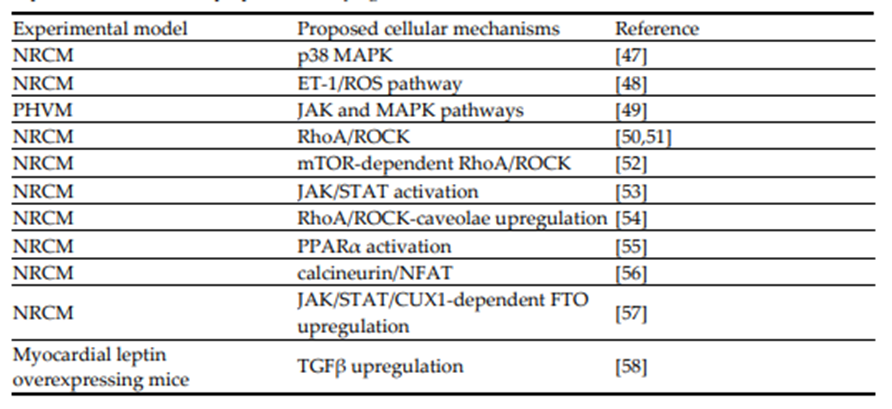
An important issue to address is the nature of adipokine contribution to risk or severity of cardiac disease, including the development of heart failure seen in obese individuals which occurs Preprints.org (www.preprints.org) | NOT PEER-REVIEWED | Posted: 27 November 2025 doi:10.20944/preprints202511.2058.v1 © 2025 by the author(s). Distributed under a Creative Commons CC BY license. 5 of 38 secondary to metabolic cardiomyopathy seen under conditions of obesity. As already noted, obesity itself has become a major world health concern. Data from the WHO show that one in eight people in the world were living with obesity in 2022 (Obesity and overweight fact sheet available from: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight). Although long considered as a major factor in heart disease development, the exact contribution of obesity is somewhat difficult to assign specifically in view of the underlying complex factors involved as well as the potential role of the obesity paradox as a complicating factor is assessing the role of obesity in cardiovascular disease. In addition, the anatomical location of increased adiposity, age of the patient and the existence of other risk factors can all ultimately contribute to the cardiovascular disease process in general [4] and, of relevance to this review, heart failure in particular. It appears that the primary heart failure subtype seen in obesity is diastolic dysfunction associated with heart failure with HFpEF [27]. In terms of fat distribution, excessive visceral fat is considered of particular importance in the development of heart failure [28] and clinical studies have shown that visceral adiposity, but not subcutaneous abdominal fat, even in the absence of heart failure was associated with concentric hypertrophic remodeling and significantly reduced left ventricular function [29]. In addition, epicardial adipose tissue may contribute to the development of heart failure via profibrotic and proinflammatory pathways [30,31]. The mechanisms by which obesity contributes to heart failure and cardiac dysfunction are varied and complex and involve defective energy metabolism and myocardial structure [32–34], disruption of autophagy [35], as well as many other metabolic and cellular defects [36,37]. Determining the role of adipokines and identifying the specific adipokines potentially involved in the development of heart failure is indeed a challenging task particularly in view of the large number of adipokines thus far identified and the diverse and indeed, opposing effects these adipokines exert on cardiovascular health [38]. As reasonably suggested by these authors, targeted interventions that affect distinct pathways would help to identify specific functions of individual adipokines affecting heart failure and potentially result in improved and novel therapeutic strategies [38].Leptin and Adiponectin and the Critical Importance of the Leptin/Adiponectin Balance When considering all adipokines thus far identified it is evident that leptin, initially identified as adipocyte-derived satiety factor by Jeffrey Friedman’s group in 1994 through cloning of the ob/ob gene [39], represents the most widely studied of these proteins not only with respect to appetite regulation and effects on energy balance but as a modulator of the cardiovascular system with respect to its influence on disease processes in general and myocardial hypertrophy and heart failure specifically. The contribution of leptin to myocardial remodelling and the proposed underlying mechanisms have been reviewed in detail by the authors and other investigators. Accordingly, this will not be comprehensively discussed here and interested readers are directed towards several recent reviews which discuss these issues [41–43]. Briefly, there is evidence that leptin, derived from either adipose, vascular or cardiac tissue contributes to the remodelling process in either a hormonal, paracrine or autocrine manner [44,45]. Leptin exerts these effects by directly acting on specific receptors which are expressed in the cardiomyocyte as well as other cardiac components such as fibroblasts. These receptors actually represent a family of six receptors termed ObR (or LepR), members of the Class 1 cytokine receptor family, which possess identical extracellular domains but which differ markedly in the composition of their intracellular domain which dictates the nature of the intracellular signalling pathway. The only exception to this is the ObRe receptor which functions as a soluble leptin binding receptor which is not anchored to the cell membrane and which is thus unable to transduce any downstream cellular signalling. The ObRb receptor which encodes the full intracellular domain likely represents the primary receptor activating pro-remodelling cell signalling pathways. Although from a general perspective, ObRb activation leads to JAK2-STAT3 activation in many cells, stimulation of non-JAK2-dependent pathways has also been documented [46]. Indeed, as summarized in Table 1 the pro-remodelling/hypertrophic effect of leptin has been linked to activation of various intracellular signalling mechanisms. Table 1. Summary of studies demonstrating pro-hypertrophic/pro-remodelling effects of leptin in various experimental models and proposed underlying molecular and cellular mechanisms.
Adiponectin, initially referred to as Acrp30 (for adipocyte complement-related protein of 30 kDa), was first identified in 1995 [59]. It is also the most abundant of the known adipokines in terms of serum concentrations and, interestingly, its plasma concentration inversely correlates with the degree of adiposity [60,61]. Adiponectin produces its effects by binding to its receptors termed AdipoR1 or AdipoR2 [62] with both receptors as well as adiponectin shown to be expressed in adult ventricular myocytes and linked to AMPK activation [63]. Indeed, AMPK activation and the resultant subsequent suppression of ERK activation, have been proposed as primary mechanisms underlying the antihypertrophic effect of adiponectin involving both AdipoR1 and AdipoR2 activation [64,65]. However, of the two receptors, AdipoR1 likely represents the primary receptor subtype underlying the antihypertrophic effect of adiponectin. For example, patients expressing an AdipoR1 mutation demonstrate hypertrophic cardiomyopathy whereas overexpression of mutant AdipoR1 in cultured cardiomyocytes or the expression of the mutant receptor in transgenic mice leads to hypertrophic responses in the absence of any other intervention [66]. These hypertrophic responses were shown to be mediated via p38/mTOR and/or ERK1/2 pathways and reversed by rapamycin [66]. Moreover, adiponectin knockout mice demonstrate an enhanced hypertrophic response to pressure overload produced by thoracic aorta banding [64]. Accordingly, adiponectin and leptin could be considered as adipokines exerting opposite effects on the cardiovascular system. As summarized in Table 2, evidence for a direct antihypertrophic effect of adiponectin is quite extensive based on studies using a variety of experimental models. Table 2. Summary of studies demonstrating anti-hypertrophic/anti-remodelling effects of adiponectin or adiponectin agonists in various experimental models and proposed primary underlying molecular and cellular mechanisms.


The studies listed in Table 2 support the concept of a salutary antihypertrophic effect of adiponectin under various experimental conditions. Interestingly, the hypertrophic response in mice subjected to pressure overload is enhanced in adiponectin null mice through a mechanism associated with enhanced activation of the prohypertrophic transcriptional factor Mef2 thus suggesting that background adiponectin is required for the induction of cardiac hypertrophy following induction of pressure overload [77]. Moreover, adiponectin deletions have been shown to exacerbate heart failure in mice subjected to thoracic aorta constriction, thus further implicating adiponectin as a protective factor [78]. 7. Omentin Omentin was first identified nearly 20 years ago as a novel glycoprotein adipokine produced in omental visceral (hence its name) adipocytes playing a potential important role in regulating the actions of insulin and thus glucose homeostasis [79]. This adipokine has two isoforms, namely omentin-1 and omentin-2 with the former demonstrating the greater abundance and biological effects [79,80]. Omentin-1 is generally considered as a beneficial agent in view of its ability to protect vascular endothelium, dilate blood vessels, improve glucose homeostasis as well as possessing antiinflammatory properties [81]. There is evidence that some of the beneficial actions of omentin-1, at least with respect to its direct hypotensive effects in normotensive rats, may be dependent on increased production of adiponectin with a resultant increased nitric oxide production [82]. With respect to myocardial remodelling and heart failure, there is emerging evidence that omentin functions as an antihypertrophic factor following myocardial insult and that it may serve as a significant predictor of development of left ventricular hypertrophy, at least in patients with Type 2 diabetes [83]. Among the first studies to demonstrate this effect was presented by Matsuo et al [84] who showed that administration of an adenoviral vector expressing human omentin to mice subjected to transverse aortic constriction or angiotensin II administration resulted in attenuation of hypertrophy and fibrosis as well as ERK phosphorylation. A similar effect was seen in cultured cardiomyocytes treated with phenylephrine in which human omentin protein effectively reduced the hypertrophic response [84]. The authors attributed the antihypertrophic effect of omentin to increased phosphorylation of AMPK. Indeed, prevention of AMPK phosphorylation prevented the antihypertrophic effect of omentin [84]. Omentin was also shown to reduce hypertrophy in another in vivo model comprising sustained coronary artery ligation. In this regard, transgenic mice expressing the human omentin gene in adipocytes demonstrated a higher survival rate, reduced hypertrophy and fibrosis 4 weeks after induction of infarction compared to their wild-type cohorts [85].Moreover, omentin-expressing transgenic mice demonstrated improved left ventricular function compared to wild-type mice as determined by echocardiography [85]. These results were also apparent in wild-type mice treated 3 days post coronary ligation with intravenous administration of adenoviral vectors expressing human omentin [85]. Interestingly, omentin-1 was also found to inhibit the prohypertrophic effect of the adipokine resistin (discussed in the following section) in cultured H9c2 cardiomyoblasts [86]. This effect was attributed to inhibition of resistin-induced upregulation of pro-inflammatory signalling involving the TLR4/MyD88/NF-κB phosphorylation pathway [86]. The clinical relevance of omentin to heart failure or myocardial remodelling remains to be determined with certainty although omentin-1 levels were shown to be decreased in elderly patients with HFpEF [87]. Similar results were reported by Huang et al [88] who demonstrated reduced plasma omentin-1 levels in patients with heart failure associated with dilated cardiomyopathy. Thus, based on these findings it has been suggested that plasma omentin-1 levels may represent a suitable marker for the development of heart failure in the clinical scenario. However, omentin-1 levels appear to be only mildly associated with left ventricular dysfunction in heart failure patients based on linear regression analyses [89].
Resistin:
The cysteine-rich adipokine resistin was first identified in 2001 and is so named due to its ability to increase insulin resistance when injected into experimental animals [90]. Of potential relevance to cardiac pathology resistin, albeit along with other proinflammatory adipokines, has been shown to increase expression of proinflammatory factors [91,92]. Resistin is expressed in the heart [93] and there is abundant evidence that resistin contributes to the myocardial remodelling process even in the absence of any pathological insult. Thus, myocardial overexpression of resistin produces a multifaceted phenotype consisting of oxidative stress, inflammation, fibrosis, apoptosis, hypertrophy and cardiac dysfunction, 9 weeks following adenoviral transfection in rats [94]. Adipose tissuespecific deletion of resistin reduced myocardial remodelling and improved cardiac function in mice subjected to pressure overload-induced aortic banding produced by 10-week thoracic aorta coarctation while, in contrast, resistin overexpression exacerbated the deleterious effect of pressure overload [95]. These effects were proposed to occur via modulation of expression of miRNAs which are important for post-transcriptional regulation of key factors involved in the hypertrophic and remodelling processes. In this particular case, pressure overload was associated with increased expression of miR148b-3p leading to DNA damage which was attenuated by resistin inhibition and increased with overexpression of the adipokine [95]. Resistin has also been implicated as a major contributor to diabetic cardiomyopathy [96] although this may reflect its ability to mediate enhanced hyperglycemia through increased insulin resistance under diabetic conditions [90]. While the above studies show that resistin can augment the remodelling response to pathological insult, there is evidence that cardiomyocyte overexpression per se can directly alter cell characteristics although this appears to vary in terms of molecular and cellular mechanisms underlying this response. Thus, adenovirus-mediated overexpression of resistin in cultured neonatal rat ventricular myocytes produced a hypertrophic response and activation of signal transduction pathways including MAPK signalling [93]. Overexpression of resistin in adult cultured rat cardiomyocytes significantly depressed cell shortening associated with defective intracellular calcium homeostasis [93]. Direct addition of resistin has been shown to produce hypertrophy in cultured H9c2 cardiac myoblasts which was attributed to inactivation of the LKB1/AMPK cell signaling pathway [97]. Moreover, direct addition of resistin to H9c2 myoblasts produced hypertrophy which was inhibited by omentin through inhibition of the TLR4/MyD88/NF-κB signalling pathway [98]. Similar opposing effects of adipokines were shown with apelin (discussed in Chapter 10), which reduced the prohypertrophic effect of resistin in H9c2 myoblasts via ERK1/2 inhibition [99]. These studies reinforce the concept that the net effect of adipokines on myocardial hypertrophy and remodelling will reflect the balance of pro- and anti-hypertrophic adipokines produced. Overexpression of resistin in NRVMs as well as in rats produced a hypertrophic response associated with AMPK inhibition and activation of mTOR and its downstream target p70S6K as well as the apoptosis signal-regulating kinase 1/c-Jun N-terminal Kinase pathway [100]. Moreover, overexpression cardiac human resistin produced right ventricular dilatation and dysfunction suggesting that resistin can contribute to right ventricular dysfunction in response to pulmonary hypertension [101]. Cardiac dysfunction associated with resistin overexpression was attributed to suppression of protein kinase A and AMP-activated protein kinase [101]. Right ventricular tissue of pulmonary hypertension patients demonstrates increased resistin expression as did right ventricular tissue from rats in which pulmonary hypertension was induced by monocrotaline treatment [101]. From a clinical perspective, serum resistin levels have been positively correlated with poor prognosis in patients with heart failure, severity of heart failure [102] as well as risk for developing heart failure in elderly subjects possibly involving the induction of proinflammatory cytokines [103,104]. This association between resistin and increased heart failure risk was also seen in younger subjects (N=2,739) as part of the Framingham Offspring Study with no relationship observed with respect to adiponectin and heart failure [105], the latter finding in contrast to other reports discussed earlier in this review. It is possible that this lack of association between adiponectin and heart failure reflects the occurrence of adiponectin resistance seen in patients with advanced heart failure [106], although a more recent study showed a clear association between serum resistin levels and incident heart failure, no association was observed in relation to subclinical myocardial fibrosis [107].
Visfatin:
Visfatin is an interesting adipokine which has undergone a number of name modifications. It was first identified in 1994 as pre-B-cell colony-enhancing factor [108]. Subsequently it was found that pre-B-cell colony-enhancing factor had a critical role in NAD biosynthesis thereby regulating the deacetylase activity of sirtuins and was thus renamed NAMPT [109]. NAMPT was found to be secreted by visceral adipocytes and renamed visfatin for “visceral fat-specific adipokine” [110,111]. To enhance clarity and maintain consistency, visfatin will be used to denote this adipokine in the present discussion. Although visceral fat tissue represents the primary source of visfatin expression and production, the protein is generally ubiquitously expressed and has been identified in cardiomyocytes and its expression can be induced by a number of factors including angiotensin II and glucose [112,113]. The expression of visfatin has been shown to be under the control of the nuclear receptor REV-ERB complex [114]. Moreover, visfatin is expressed in cardiac fibroblast which may be of relevance in terms of its role in myocardial remodelling [115], as discussed further below. In clinical studies, plasma visfatin levels have been shown to be associated with an increased incidence of major adverse cardiovascular events in myocardial infarction patients [116]. Studies vis a vis cardiac effects of visfatin suggest it being a beneficial adipokine as initially shown in terms of its ability to produce a hypoglycemic effect under obesity or type 2 diabetes [117]. Moreover, studies have shown that visfatin administration to mice subjected to myocardial infarction followed by reperfusion reduces infarct size in these animals [118]. These effects may be due to visfatin-induced increased NAD and ATP production in the cardiomyocyte leading to increased cell survival via a reduction in apoptosis and stimulation of autophagy [119]. Further evidence for a beneficial role of visfatin comes from studies demonstrating an increased extracellular monocytederived circulating visfatin levels in pressure overloaded mice subjected to thoracic aortic banding [120]. Importantly, pharmacological inhibition of visfatin by FK866 increased cardiomyocyte apoptosis and cardiac decompensation in these animals [120]. While stimulation of autophagy is considered a potential mechanism for reduced postinfarction remodelling, visfatin has paradoxically been shown to directly stimulate collagen synthesis and proliferation of cultured cardiac fibroblasts through a mechanism involving p38MAPK, PI3K, and ERK 1/2 pathways thus potentially contributing to enhanced myocardial remodelling [121]. Indeed, transgenic mice overexpressing visfatin demonstrated pronounced spontaneous cardiac hypertrophy 6 months after treatment in the absence of other prohypertrophic interventions [122]. The hypertrophic response was associated with various intracellular signalling pathways including activation of mitogen-activated protein kinases JNK1, p38, and ERK as well as increased calcineurin levels and NFAT translocation to nuclei [122]. A prohypertrophic effect of visfatin has also been demonstrated in a study using H9c2 fibroblasts where exposure of these cells for up to 72 hours resulted in a hypertrophic response proposed to be mediated via an endoplasmic reticulum stress and transient TRPC1 pathway [123]. Visfatin administration has also been found to increase cardiomyocyte hypertrophy, increase cardiac fibrosis and reduce cardiac function when administered to mice subjected to thoracic aorta banding [124]. These deleterious effects of visfatin were found to be associated with a macrophage-dependent increase in oxidative stress [124]. Evidence for a deleterious role of visfatin was presented in a recent study in which a visfatin neutralizing antibody reduced remodelling and improved cardiac function in rats subjected to a sustained 4-week myocardial infarction via inhibition of the visfatin/TLR4 inflammatory cascade [125]. It has also been proposed that visfatin may play a beneficial role in in mediating reverse myocardial remodelling seen following debanding of aortic banded mice or in patients with aortic stenosis following aortic valve replacement [126]. This concept was suggested based on myocardial visfatin measurements which were significantly reduced both during aortic banding in mice or stenosis in patients whereas values increased following debanding or aortic valve replacement [126]. Although this study provides solely indirect evidence the authors suggest that visfatin may be an important regular of myocardial remodelling due to pressure overload. Moreover, visfatin upregulation has been shown to protect the heart against the development of cardiomyopathy in a high fat diet-induced mouse diabetes model consisting of a mechanism involving an NAD+ - dependent anti oxidative stress process [127]. As noted above, visfatin is also expressed in cardiac fibroblasts and upregulated, along with Type 1 procollagen under high glucose (30 mmol/l compared to 5.5 mm/l for control conditions) exposure through a mechanism involving the ROCK signalling pathway [115]. Although exposure of fibroblasts to high glucose conditions enhanced fibroblast proliferation, a potential role of visfatin was not studied [115]. When taken together, it apparent that visfatin plays complex and multifaceted roles in the development of heart failure, possibly dependent on the experimental model. This has been strongly suggested in a study using mice subjected to thoracic aortic banding, i.e. pressure overload, where it was shown that endogenous visfatin protects the heart in this model although its cardiac-specific overexpression enhances myocardial hypertrophy, fibrosis, apoptosis, mitochondrial energy dysfunction and reduces cardiac function as assessed by echocardiography [128]. Loss of cardiac visfatin per se in the absence of any other insult, produces a heart failure phenotype consisting of metabolic derangements, myocardial remodelling and sudden cardiac death which were dependent on NAD+, thereby suggesting a critical role for endogenous visfatin for the maintenance of normal cardiac function [129].
Apelin:
The adipokine peptide apelin was first identified in the bovine stomach [130] and subsequently shown to exert its effects through the activation of the g-protein coupled APJ receptor. First identified in 1993, this receptor is interesting in that it shares approximately a 50% homology with the AT1receptor [131] although it is not activated by angiotensin II. Indeed, APJ activation negatively regulates AT1 receptor activation through negative allosteric regulation [132] and apelin has been shown to exert effects opposite to those seen with angiotensin II and protect again angiotensin IIinduced cardiac fibrosis [133]. Although apelin is generally considered as an antihypertrophic factor, as discussed below, there is evidence that the APJ receptor per se may function as a dual receptor both promoting and inhibiting the hypertrophic response. In this regard, Scimia and coworkers [134] have shown that APJ knockout mice demonstrate a reduced hypertrophic response in response to sustained thoracic aorta binding whereas mice lacking apelin retain the hypertrophic response thus suggesting an apelin-independent role of APJ. The authors’ primary conclusion invokes a stretchinduced activation of the APJ resulting in a hypertrophic response via a β-arrestin dependent mechanism whereas apelin confers an APJ-dependent antihypertrophic effect by activation of Gprotein Gαi [134]. Overall, APJ appears to play diverse roles in cardiac intracellular and the receptor has been shown to function as a sensor in response to myocardial stretch resulting in a positive inotropic response [135]. Apelin is initially secreted as a 77-amino acid preproprotein which is subsequently cleaved to shorter fragments. The most abundant and important of these fragments in terms of biologic effects is the 13-amino acid apelin-13 fragment which has been identified both in plasma [136] as well as human heart [137]. Recently, a second endogenous ligand for the APJ receptor named Elabela, a bioactive micropeptide, has been identified and appears to play important roles in numerous physiological processes [138]. Elabela has been shown to exert numerous cardiobeneficial effects including reduction in oxidative stress-induced injury [139] as well as a reduction in pressure-overload induced cardiac dysfunction and hypertrophy produced by angiotensin II infusion in mice [140]. Elabela has also been reported to improve cardiac function and reduce hypertrophy in heart failure produced by myocardial infarction in mice through a mechanism involving vascular endothelial growth factor receptor 3 activation [141]. While the beneficial results of Elabela are somewhat predictable based on the ability of apelin to reduce hypertrophy and heart failure via APJ activation, it is interesting that some differences between Elabela and apelin have been reported. One such area involves the enzyme ACE2, a homolog of ACE1 and a negative regulator of the RAS by cleaving angiotensin 2 and thus generally considered as an inhibitor of myocardial remodelling [142]. Apelin has been shown to increase ACE2 levels in the failing heart and to couple apelin to RAS [143] and it is interesting to add that the activation of the Apelin-ACE 2 pathway has been proposed as a mechanism for the beneficial effects of the SGLT2 inhibitor canagliflozin in a model of hypertrophy and heart failure in saltsensitive rats [144]. This effect of apelin also reveals differences between apelin and Elabela. For example, when comparing the effects of apelin and Elabela in the study just cited [140], it was shown that apelin treatment did not modulate ACE mRNA expression but upregulated ACE2 expression in pressure-overloaded hearts. In contrast, Elabela did not affect ACE2 expression but downregulated ACE expression. Thus, while both AJP agonists reduced the effect of pressure overload equally, this likely was mediated by different effects on the angiotensin system [140]. Studies into the cardiac influence of apelin have primarily, but not exclusively (see below) demonstrated beneficial effects including reduction is ischemic and reperfusion injury [145], reduction in blood pressure through nitric oxide generation [146], improved myocardial repair following myocardial infarction [147,148], increased myocardial vascular density in a mouse model of diabetic cardiomyopathy [149], as well as attenuation of atrial fibrosis which can contribute to the development of atrial fibrillation [150]. Thus, targeting the apelin system is considered as a potentially effective approach towards mitigating a number of cardiovascular disorders including cardiac hypertrophy [151,152]. In terms of heart failure, cardiac apelin expression has been shown to be reduced in experimental left ventricular hypertrophy and heart failure comprising Dahl saltsensitive rats which was reversed by treatment with the AT1receptor blocker telmisartan [153].Moreover, apelin knockout has been shown to produce impaired contractility and a hypertrophy phenotype in aging mice in the absence of any other intervention [154]. Although these studies point to depressed apelin levels in the hypertrophied myocardium, plasma levels are increased as demonstrated in both animal and human studies, which the authors attributed to a compensatory mechanism aimed at blunting the hypertrophic response [155]. However, hypertensive patients with left ventricular hypertrophy were shown to exhibit reduced plasma apelin levels when compared to hypertensive subjects without left ventricular hypertrophy [156]. The differences between myocardial vs plasma apelin levels have been attributed to the fact that failing hearts release apelin into the circulation in contrast to apelin uptake by non-failing hearts [157]. Apelin can also produce beneficial effects through its positive inotropic influence [158,159] as well as afterload reduction [159] and clinical studies have shown that apelin-13 infusion improves cardiac function in heart failure patients [160,161]. Apelin also has been shown to reduce severity of acute heart failure rabbits produced by sodium pentobarbital administration through a mechanism involving reduction in endoplasmic reticulum stress [162] whereas a selective APJ receptor agonist improved indices of cardiac function in the renal hypertensive rat model of cardiac hypertrophy and reduced cardiac output [163]. In addition to the beneficial effects of apelin-13, a metabolically resistant analogue of apelin-17 has been shown to reduce myocardial remodelling and improve ventricular function in a murine model of heart failure produced by sustained coronary artery ligation [164]. Of particular relevance to obesity, apelin has been shown to reverse the direct hypertrophic effect of a 24-week high fat diet in mice when administered for two weeks following high fat diet cessation [165]. This effect was associated various improved indices including cardiac function and improved intracellular Ca2+ homeostasis, endoplasmic reticulum stress as well as autophagy [165]. Moreover, apelin prevents the development of heart failure in mice fed a high fat diet and subjected to pressure overload via aortic binding [166]. These effects of apelin were associated with improved myocardial energy metabolism as evidenced by improved fatty acid utilization and glucose oxidation as well as reduced mitochondrial injury [166]. Apelin can also reduce myocardial remodelling by a reduction in fibroblast proliferation. Indeed, treatment of mouse cardiac fibroblasts with apelin reduced fibroblast activation and collagen production which was mediated by a reduction in sphingosine kinase 1, which was similarly observed in an in vivo model of heart failure produced by aortic banding [167]. Other mechanisms by which apelin could reduce the hypertrophic response to various factors is likely multifactorial but the antihypertrophic effect has been shown to be related to reduced oxidative stress by increased myocardial catalase activity [168] as well as a potential reduction in the inflammatory response as demonstrated by a reduction in inflammation-associated markers in cultured cardiomyoblasts [169]. Based on numerous positive results using apelin in heart failure models, the peptide shows promise as a potential therapeutic agent although its effectiveness is limited by a short in vivo halflife of less than 5 minutes [170]. This limitation can be overcome by developing stable analogues of apelin which are not readily degradable or by identifying novel delivery strategies. For example, with respect to the latter it has previously been shown that apelin encapsulation in liposome nanocarriers conjugated with PEG polymer produced sustained apelin release and improved cardiac function in a mouse model of pressure-overload induced heart failure produced by14-day thoracic aortic banding [171]. As briefly noted above, although cardiac antihypertrophic effects of apelin have been extensively demonstrated, contrary findings have also been reported. Thus, the same group has consistently demonstrated that apelin can induce hypertrophy in cultured H9c2 rat myoblasts by stimulating autophagy pathways through mechanisms associated with increased PI3K phosphorylation [172,173] as well as caveolin-1 inhibition [174] and increased endoplasmic reticulum stress [175]. These findings add substantial complexity to our understanding of apelin and cardiac hypertrophy although it should be appreciated that myoblasts may respond differently to various stimuli when compared to effects on ventricular myocytes.
Vaspin:
The 47 kDa 415 amino acid adipokine vaspin (so named from visceral adipose tissue-derived serpin) was first identified in 2005 in visceral adipose tissue of obese and Type 2 diabetic rats and proposed to function beneficially to reduce insulin resistance [176]. Our current knowledge of the relevance of vaspin to cardiac health is somewhat limited although low plasma concentrations have been shown to be associated with increased major cardiac events, including heart failure, in patients following acute myocardial infarction [177]. A small number of studies have investigated the effect of vaspin on cardiac hypertrophy experimental models with uniform findings showing beneficial effects in terms of reducing hypertrophy and myocardial remodelling. For example, vaspin was shown to supress myocardial fibrosis and improve cardiac function in mice subjected to heart failure produced by either myocardial infarction, thoracic aortic banding or angiotensin 2 infusion, each treatment for a period of four weeks [178]. These beneficial effects were attributed to a reduction in oxidative stress subsequent to suppression of the PI3K/Akt pathway [178]. A more recent study showed that vaspinknockout mice demonstrate a greatly enhanced myocardial remodelling and hypertrophy as well as Preprints.org (www.preprints.org) | NOT PEER-REVIEWED | Posted: 27 November 2025 doi:10.20944/preprints202511.2058.v1 © 2025 by the author(s). Distributed under a Creative Commons CC BY license. 13 of 38 ventricular dysfunction produced by 9-day isoproterenol administration thereby further demonstrating an antihypertrophic effect of vaspin [179]. Based on these studies as well as studies using cultured neonatal cardiomyocytes, the authors demonstrated a key role for enhanced PI3K/AKT/mTOR pathway-dependent autophagy as principally accounting for vaspin-induced protection [179]. Interestingly, vaspin has also been shown to reduce cardiomyopathy and myocardial remodelling in a mouse model of lipoatrophy via stimulation of AKT phosphorylation [180].
Adipsin :
The discovery of the adipokine adipsin, also known as complement factor D, in 1987 predates the identification of leptin as a protein produced by adipose tissue [181]. Adipsin plays an important role in the complement system primarily by serving as the rate-limiting factor in the alternative complement pathway [182]. In addition, adipsin is important in the regulation of pancreatic insulin release by promoting insulin release [183] via the C3a/C3a receptor 1 [Complement C3a receptor 1 or C3aR1] pathways well as protecting pancreatic beta cells under diabetic conditions [reviewed in 184]. Although adipsin has been implicated as a potential contributor to various cardiovascular disease including those associated with vascular dysfunction [reviewed in 185], at present there is a paucity of data related to its potential role in myocardial remodelling or the development of heart failure. Substantial studies need to be carried out in order to elucidate the potential role of this adipokine in this form of pathology.
Asprosin:
Asprosin is a relatively novel adipokine first discovered in 2016 as a glucogenic hormone which is recruited primarily to the liver where it produces rapid glucose release through the activation of the G-protein dependent cAMP/PKA pathway [186]. Injection of asprosin to mice results in rapid increase in blood glucose suggesting that targeting the asprosin pathway could be therapeutically effective towards the treatment of diabetes mellitus [186]. Asprosin expression has been identified in rat cardiomyocytes and found to be decreased along with serum levels of the adipokine in animals subjected to streptozotocin-induced diabetes [187]. In terms of cardiac remodelling and heart failure, one clinical study has suggested that enhanced asprosin plasma levels may be associated with a reduction in adverse events in patients with dilated cardiomyopathy [188]. Based on studies using hypoxic cultured cardiac myoblasts treated with asprosin, the authors attributed the beneficial effects to increased cell viability concomitant with improved mitochondrial function [188]. The use of SGLT-2 inhibitors such as empagliflozin for the treatment of heart failure patients has been shown to be associated with elevated serum asprosin levels suggesting that asprosin may account, at least in part, to the beneficial effects of these drugs [189]. While the findings noted above suggest a beneficial role of asprosin, a recent report demonstrated that circulating asprosin concentrations are increased in heart failure patients and that elevated levels of this adipokine were positively correlated with the incidence of heart failure leading the authors to suggest that elevated asprosin may be a risk factor related to the development of heart failure [190]. This finding is supported by the observation that elderly patients with Type 2 diabetes and left ventricular diastolic dysfunction exhibited elevated asprosin serum levels when compared to patients without left ventricular dysfunction [191]. Moreover, increased serum levels of asprosin were associated with increased risk of developing left ventricular dysfunction in these patients [191].
Chemerin:
Chemerin, which is also known as tazarotene-induced gene 2 was originally identified in 1997 in psoriatic skin lesions [192] and proposed to function as an immunomodulator and to play an important role in a number of pathologies [193,194]. Although chemerin expression has been Preprints.org (www.preprints.org) | NOT PEER-REVIEWED | Posted: 27 November 2025 doi:10.20944/preprints202511.2058.v1 © 2025 by the author(s). Distributed under a Creative Commons CC BY license. 14 of 38 identified in various tissues including visceral adipose tissue, placenta and liver, it has also been identified in the heart [195]. In terms of heart disease, serum chemerin levels are elevated in patients with coronary artery disease suggesting that this adipokine could serve as a useful marker and predictor for coronary disease and possibly other cardiovascular events [196]. Moreover, serum chemerin levels have been shown to be a predictor of adverse cardiac events in patients with heart failure [197]. Its role in the development of heart failure and myocardial remodelling is at present unknown although at least one of the three receptors involved in chemerin cell signalling, CCRL2, has been identified in the heart and shown to participate in experimental autoimmune myocarditis in mice [198]. Although substantial future research is necessary in order to fully delineate the role chemerin in cardiac pathology, including myocardial hypertrophy and heart failure, there is the potential of targeting the chemerin system as a therapeutic strategy for a number of cardiovascular pathologies [199].
Meteorin-like protein Metrnl is so named as it shares substantial amino acid sequence homology with the CNS-specific neurotrophic factor Meteorin [200]. Metrnl has been shown to play important roles in immunology, inflammation and metabolism such as improved glucose metabolism [200]. As such, it likely contributes to a variety of pathologies and may serve as a target for therapeutic interventions or as a treatment modality for various conditions such as muscle regeneration [200,201]. Unlike Meteorin, Metrnl is ubiquitously expressed in a variety of tissues [202] and has been shown to be highly expressed in cardiac tissue [202,203] and to demonstrate cardioprotective and antiremodelling properties in a number of experimental settings. For example, with respect to cardioprotection, Metrnl has been implicated in cardioprotection due to enhanced angiogenesis in mice subjected to acute myocardial infarction [204]. These effects were shown to be mediated by activation of stem cell factor receptor KIT resulting in increased endothelial cell population in the infarct border zone [204]. While this study does not assign a direct antihypertrophic effect to Metrnl, it demonstrates a potential to reduce heart failure following infarction by enhancing cardiac repair. Moreover, Metrnl overexpression in cardiac macrophages resulted in substantial cardioprotection in mice subjected to myocardial ischemia and reperfusion, which reduced Metrnl expression on its own [205]. These protective effects were proposed to be mediated by increased macrophage polarization from M1 to M2 through the activation of AMPK phosphorylation resulting in diminished release of proinflammatory factors [205]. Metrnl has also been shown to reduce cardiotoxicity in mice treated with the antitumour agent doxorubicin (Adriamycin). In this regard, cardiac-specific overexpression of Metrnl reduced oxidative stress, apoptosis and cardiac dysfunction in these animals and improved survival while not affecting the antitumour properties of doxorubicin [206]. These beneficial effects were attributed to Metrnl-induced SIRT1 activation via the cAMP/PKA pathway [206]. SIRT1 upregulation has also been shown to mediate the antihypertrophic effect seen in Metrnl overexpressing mice subjected thoracic aorta coarctation [207]. Metrnl is highly expressed in cardiac myocytes and its expression has been shown to be dependent on PPARα activity [203]. Importantly, genetically generated Metrnl deficient mice demonstrated enhanced adverse myocardial remodelling as demonstrated by increased hypertrophy, fibrosis, pro-inflammatory markers as well as defective left ventricular function following 7-day isoproterenol infusion, effects which were reversed by Metrnl overexpression [203]. Although the precise mechanism underlying the antihypertrophic effect of Metrnl is unknown and requires further studies, these authors have demonstrated a potential role of p38-MAPK activation with subsequent phosphorylation of the transcription factor CREB leading to the upregulation of the protective factor PGC1α [203]. Another potential cellular process by which Metrnl could protect against myocardial remodelling is by targeting autophagy. For example, in both Type 1 and Type 2 diabetes mouse models, cardiomyopathy produced by hyperglycemic conditions was attenuated in animals with cardiac Metrnl overexpression whereas Metrnl depletion exacerbated this condition [208]. These Preprints.org (www.preprints.org) | NOT PEER-REVIEWED | Posted: 27 November 2025 doi:10.20944/preprints202511.2058.v1 © 2025 by the author(s). Distributed under a Creative Commons CC BY license. 15 of 38 beneficial effects were associated with increased autophagy through stimulation of ULK1, a key factor in autophagy initiation [209]. While this study implicates enhanced autophagy as a mechanism of protection against diabetic cardiomyopathy, protection again postinfarction myocardial remodelling by Metrnl was found to be associated with reduced autophagy. In this regard, cardiac Metrnl overexpression significantly reduced myocardial remodelling seen in hypertensive rats via autophagy inhibition and Metrnl administration reduced angiotensin 2-induced hypertrophy in cultured H9c2 myoblasts via BRCA2/Akt/mTOR signalling [210]. These effects were associated with reduced autophagy. Indeed, similar results were obtained in a mouse infarct model in which the subsequent myocardial remodelling was attenuated by myocardial Metrnl overexpression through a mechanism mediated by reduced autophagy [211]. Thus, inhibition of autophagy by Metrnl may represent a common mechanism for the antiremodelling effect of this adipokine. The protective effect of Metrnl on myocardial remodelling and heart failure appears to be borne out in some clinical studies in that low plasma levels of this adipokine have been found to be associated with increased severity of heart failure in elderly patients [212]. However, post infarction clinical studies suggest a potential deleterious role of this adipokine. Thus, an assessment of 400 patients with newly diagnosed heart failure revealed a higher incidence of all cause and cardiovascular death associated with elevated serum Metrnl levels [213]. The reason for the apparent discrepancy is uncertain, but as suggested by others [203], the elevated plasma levels of this adipokine may reflect an attempted compensatory protective mechanism aimed at mitigating the heart failure process rather than a contributor to the pathology. Clearly, substantial further studies are necessary in order to clarify this issue and indeed the overall contribution of Metrnl to the heart failure process.
Progranulin:
The 539 amino acid glycoprotein progranulin, first isolated from a tumorigenic cell line [214,215] is produced by various cell types but confirmed rather recently as an adipokine exerting a plethora of biological effects and participating in numerous diseases particularly those involved in neural function [216]. It exerts anti-inflammatory properties, inhibits neutrophil activation, reduces oxidative stress [217] and has been shown to protect the ischemic and reperfused heart [218]. Although limited in scope, there is emerging evidence that progranulin inhibits myocardial remodelling and heart failure. From a general perspective, the ability of progranulin to limit the inflammatory response can bestow significant benefit in terms of attenuating cardiovascular pathology [219]. A specific contribution for this antiremodelling effect may be dependent on the ability of progranulin to reduce infarct size as demonstrated when administered to mice subjected to sustained coronary artery occlusion [220], based on the fact that reduced infarct size would attenuate subsequent myocardial remodelling. Progranulin is abundantly expressed in mouse cardiac macrophages whereas progranulin deficient mice exhibited higher rates of mortality, increased left ventricular fibrosis and increased arrhythmogenesis following 14-day coronary artery ligation which was associated with alterations in macrophage-derived cytokine production, namely higher levels of TGF-β, IL-4R, and lower amounts of IL-1β, IL-10 [221]. Moreover, in the absence of pathological insult, progranulin deficiency has been shown to enhance age-depended cardiac hypertrophy as reported for 18-month-old mice via a mechanisms involving complement C1q and activated βcatenin protein expression [222], key components of the Wnt signalling pathway which can contribute to cardiac hypertrophy [223]. Indeed, blocking this pathway attenuated the cardiac senescence-associated increased hypertrophic response [222]. From patient studies, it is interesting to note that enhanced progranulin levels 7 days following acute myocardial infarction were associated with improved left ventricular function as assessed 6 months following infarction [224]. While suggestive of a salutary role of progranulin following myocardial infarction this study has limitations in terms of small patient population studied (18 subjects), lack of evidence for a cause-and-effect relationship or mechanistic insights into the apparent benefit on left ventricular function.
Neuroregulin :
Nrg4 is an adipokine first identified in 1999 [225] and is a member of the epidermal growth factor family which is widely expressed in brown adipose tissue as well as in many other tissues [226,227]. Nrg4 exerts its effects via the activation of the ErbB4 receptor tyrosine kinases and has been shown to participate in numerous physiological and pathophysiological processes [226,227]. There is recent emerging evidence that Nrg4 exerts cardioprotective effects as has been shown in diabetes models of cardiomyopathy [228,229]. Moreover, antihypertrophic and antiremodelling effects of Nrg4 have been reported. For example in a mouse model of 14-day isoproterenol infusion, myocardial remodelling was attenuated by a subsequent four-week Nrg4 treatment [230].This effect of Nrg4 was associated with reduced hypertrophy, inflammatory response, fibrosis as well as apoptosis and improved cardiac function [230]. These salutary effects were found to be dependent on the AMPK/NF-κB pathway [230]. Moreover, overexpression of Nrg4 attenuated myocardial fibrosis and cellular apoptosis in spontaneously hypertensive rats [231]. Using AC16 cardiomyocytes (a human ventricular myocyte cell line), Zhang et al [232] recently reported that levels of Nrg4 can be increased following its stabilization by the RNA-binding protein Poly[RC] Binding Protein 2 thus resulting in reduced hypertrophy, oxidative stress and inflammatory response following angiotensin 2 administration. Nrg4 exerts a myriad of beneficial effects which could be conducive to indirectly limiting myocardial remodelling including antioxidant and anti-inflammatory properties as well as reducing atherosclerosis [230,232,233]. Moreover, clinical observation showed that serum Nrg4 levels are inversely correlated with the severity of coronary artery disease [234].The results thus far concerning Nrg4 have been promising in identifying a protective batokine limiting myocardial remodelling and heart failure although substantial future work is required to fully delineate the role of Nrg4 in the heart failure process.
Retinol Protein Binding:
RBP4 is an adipokine and transporter protein which exerts a variety of functions including transport of vitamin A into the circulation. RBP4 generally is associated with deleterious effects including increased insulin resistance primarily by its ability to stimulate pro-inflammatory cytokines thus contributing to the development of type 2 diabetes [235]. RBP4 is emerging as an important contributor to various pathologies related to cardiovascular disease including promoting oxidative stress, apoptosis, alterations in lipid metabolism as well as increasing insulin resistance as already alluded to [reviewed in 236]. Aside from its numerous effects promoting cardiac pathology there is some evidence that RBP4 promotes cardiac hypertrophy through a number of cell signalling processes. For example, Gao and coworkers showed that RBP4 directly induces hypertrophy when added to mouse neonatal ventricular myocytes, an effect associated with increased expression of proinflammatory and oxidative stress markers [237]. The authors attributed the prohypertrophic effect of RBP4 to activity of the TLR4/MyD88 pro-inflammatory pathway [237]. Although concrete clinical data are currently lacking, there is evidence that elevated levels of RBP4 are a predictor of adverse cardiac events in elderly patients with heart failure [238] or the development of cardiomyopathy in diabetic individuals [239].
CTRP family:
CTRPs represent a family of adiponectin paralogues exerting proinflammatory effects which have been shown to modulate a myriad of conditions including cardiovascular disease such as coronary artery disease via their ability to modulate the development of atherosclerosis [reviewed in 240, 241]. However, the complexity of their actions is underscored by the fact that at least 15 of these proteins have been identified possessing diverse biological effect including diverse effects on myocardial remodelling and heart failure acting through various cell signalling processes. A number Preprints.org (www.preprints.org) | NOT PEER-REVIEWED | Posted: 27 November 2025 doi:10.20944/preprints202511.2058.v1 © 2025 by the author(s). Distributed under a Creative Commons CC BY license. 17 of 38 of these proteins have been studied in terms of their ability to modulate the myocardial disease and the remodelling process in particular.
CTRP1:
CTRP1 has not been extensively studied vis a vis myocardial remodelling although current evidence suggests that it may contribute to myocardial remodelling by its ability to increase cardiac fibrosis. For example, CTRP1-deficient mice exhibited improved survival, reduced oxidative stress and reduced inflammation following induction of myocardial infarction by supressing macrophage activation through enhancing interaction between the adiponectin AdipoR1 and TLR4 [242]. In contrast, mice receiving CTRP1 injection exhibited worse outcomes following infarction [242]. On the other hand, CTRP1 deficiency has been shown to exacerbate angiotensin II-induced cardiac hypertrophy, fibrosis, inflammation and oxidative stress associated with diminished cardiac function in mice through the activation of the AMPKα pathway [243]. As with many of the identified adipokines, contrary findings implicating CTRP1 as a proremodelling factor have also been reported through its ability to increase fibrosis. In this regard, mouse fibroblasts treated with recombinant CTRP1 demonstrated increased release of proinflammatory factors whereas fibroblasts co-cultured with macrophages treated with recombinant CTRP1 demonstrated increased proliferation, effects attributed to activation of the NADPH oxidase 2/p38 pathway in macrophages [244].
CTRP3:
CTRP3 has been shown to protect the ischemic and reperfused heart [245] and more recently to exert an antihypertrophic effect in mice subjected to thoracic aortic constriction. In this regard, CTRP3 knockout mice demonstrated enhanced hypertrophy and reduced left ventricular function 4 weeks post aortic coarctation whereas those animals overexpressing CTRP3 showed diminished cardiac hypertrophy and fibrosis and improved left ventricular function [245]. The beneficial effects of CTRP3 overexpression was attributed to inhibition of the p38/CREB pathway and a reduction in p38-induced endoplasmic reticulum stress [246]. Similar antihypertrophic effect of CTRP3 we also observed in an identical model which was accompanied by improved mitochondrial function and reduced oxidative stress [247]. These beneficial effects of CTRP3 overexpression were attributed to activation of the UPRmt response, a mechanism likely mediated by improved protein folding [247]. CTRP3 overexpression has also been shown to limit diabetic cardiomyopathy in rats produced by streptozotocin administration or myopathy secondary to lipopolysaccharide-induced sepsis in mice through mechanisms involving activation of the AMPKα pathway [248,249]. Thus, overall evidence suggests that CTRP3 exerts an antihypertrophic and antiremodelling effect in various experimental models of heart failure. A contribution to the beneficial effect of CTRP3 may also reflect its ability to inhibit fibrosis. Indeed, this has been demonstrated in rats subjected to myocardial infarction as well as cultured cardiac fibroblasts where CTRP3 overexpression attenuated myofibroblast differentiation and extracellular matrix production through AMPK-dependent mechanisms resulting in inhibition of Smad3 activation myofibroblast differentiation [250]. It is noteworthy however that opposite effects of CTRP3 have also been reported. Thus, overexpression of CTRP3 resulted in enhanced myocardial hypertrophy and heart failure following 4-week thoracic aortic binding in mice by increased activation of the TAK1-JNK pathways whereas CTRP3 knockdown exerted beneficial effects by inhibiting these signalling pathways [251].
CTRP6:
Although there is a paucity of published data, there is evidence that CTRP6 exerts a beneficial effect in reducing heart failure. Heart failure produced by 2 weeks of isoproterenol injection in mice was attenuated by CTRP6 overexpression as demonstrated by improved cardiac function, reduced apoptosis and mitochondrial reactive oxygen species through a mechanism mediated by the AMPK Preprints.org (www.preprints.org) | NOT PEER-REVIEWED | Posted: 27 November 2025 doi:10.20944/preprints202511.2058.v1 © 2025 by the author(s). Distributed under a Creative Commons CC BY license. 18 of 38 pathway [252]. As these effects were associated with diminished myocardial injury per se, it is possible that the beneficial effect was secondary to a cardioprotective influence of CTRP6, and not a direct antihypertrophic/antiremodelling effect of the protein. However, CTRP6 has been recently show to have an antiremodelling effect in a rat infarction model primarily by its direct ability to attenuate fibroblast migration thus contributing to an antifibrotic effect [253]. This influence of CTRP6 was attributed to AMPK and Akt activation targeting the RhoA/MRTF-A pathway [253].
CTRP9:
Few studies have been reported concerning CTRP9 with diverse implications on its role in myocardial remodelling or heart failure. In a rat model of right ventricular failure produced by sustained pulmonary artery banding, CTRP9 was shown to exert a protective effect by decreasing the production of reactive oxygen species and apoptosis in the right ventricle [254].These effects were attributed to CTRP9 induced AMPK-dependent transcriptional activation of antioxidant enzymes, effects dependent also on adiponectin AdipoR1 and AdipoR2 receptor activation [254]. A beneficial effect of CTRP9 was further demonstrated in a model of cardiac hypertrophy in mice produced by administering a high fat diet of 26 weeks. These animals developed cardiac hypertrophy and fibrosis as well as endoplasmic reticulum stress-induced apoptosis which were enhanced by CTRP9 knockout as was left ventricular dysfunction, the latter being absent in wild type mice [255].The authors proposed the LKB1/AMPK signalling pathway as the primary cellular mechanism underlying the beneficial effects of TRP9 [255]. In contrast to the beneficial effects reported, as noted above, Appari et al have shown that CTRP9 overexpression exacerbates myocardial remodelling and enhanced cardiac dysfunction in a mouse model of pressure overload produced by thoracic aortic banding, whereas CTRP9 knockout mice were protected with a resulting diminished myocardial remodelling response [256]. From a mechanistic perspective, the authors implicated enhanced activated prohypertrophic ERK5 as the primary mechanism underlying the prohypertrophic effects of CTRP9 in response to pressure overload [256].
CTRP12 :
Although only one study has been reported, the findings suggest that CTRP12 protects against myocardial remodelling and the development of heart failure. Thus, CTRP12 overexpression reduced cardiac apoptosis, oxidative stress, inflammation and improved left ventricular function in rats subjected for 6 weeks to myocardial infarction produced by coronary artery ligation, whereas CTRP12 deletion enhanced remodelling and worsened left ventricular dysfunction [257]. The salutary effects of CTRP12 overexpression were associated with activation of the TAK1-MAPK-JNK pathway in the protected hearts post-infarction [257].
CTRP15:
Although CTRP15 has not been extensively studied, there is evidence, albeit limited that it exerts anti-remodelling effects. Thus, cardiac overexpression of CTRP15 in mice subjected to pressure overload-induced heart failure produced by sustained thoracic aortic banding resulted in reduced myocardial remodelling and improved cardiac function [258]. The beneficial effect of CTRP15 was attributed its direct antifibrotic effect, as demonstrated in cultured cardiac fibroblasts [258]. Moreover, the antifibrotic effect of CTRP15 was shown to be mediated by IR/IRS-1/Akt pathwaydependent Smad3 activation [258]. Thus, in closing it is clear that many members of the CTRP family exert profound effects on myocardial remodelling and heart failure with a majority of these proteins demonstrating beneficial effects, albeit with some discrepant results. These are summarized in Figure 2.
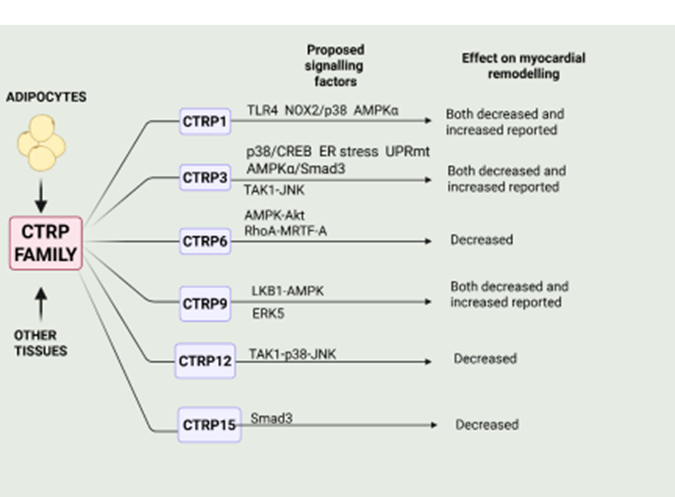
Figure 2: Summary of primary effects of members of some members of the CTRP family and the cell signalling factors implicated in these actions. Those factors exerting a protective effect are shown above the arrow whereas those factors below the arrow depict those factors mediating adverse effects of myocardial remodelling for the respective CPRT protein. Please refer to text for details. Created by BioRender.com.
Summary, Overall Conclusions and Future Directions :
The relationships between adipokine production and cardiac diseases including those involving myocardial remodelling and heart failure are complex and difficult to delineate. Among the reasons for such complexity is that many of these adipokines exert both beneficial and deleterious cardiac effects as discussed in this review, effects occurring via multifaceted molecular and cell signalling mechanism. Moreover, as is evident from the present discussion, a multiplicity of cell signalling processes have been implicated for individual adipokines. While many adipokines exert prohypertrophic and pro-remodelling cardiac effects, at the same time a large number of these proteins are considered as potential treatments for cardiovascular disorders [259]. In a recent extensive review, Packer outlined the role of adipokines in the context of their overall contribution to HFpEF providing evidence for an adipose-driven disorder which dominates in importance when compared with the contribution of hypertension [260,261]. In his detailed and compelling discussion [260] Packer assigns adipokines to three specific categories: Domain I consisting of adipokines secreted primarily by healthy adipocytes that act as cardioprotective agents (e.g. adiponectin, omentin, Nrg4, CTRP3, CTRP9); Domain II adipokines secreted primarily by visceral adipocytes and which also exert cardioprotective and antiremodelling effects (e.g. vaspin, nesfatin, apelin, adipsin, progranulin) and lastly, Domain III adipokines are synthesized primarily by hypertrophied and proliferating adipocytes seen in obese individuals which contribute to cardiac inflammation and hypertrophy resulting in heart failure with HFpEF (e.g. chemerin, leptin, resistin) [260]. While this concept pertains primarily to HFpEF the concept of selective and specific modulation of cardiac hypertrophy by adipokines can likely be applied to other cardiac pathologies. In view of the plethora of adipokines thus far identified particularly those affecting the cardiac hypertrophic and remodelling processes, substantial further research is required to fully appreciate the contribution of specific adipokines to heart failure. What is clear is that a balance between beneficial and deleterious adipokines will determine their contribution to cardiac pathology including heart failure. This concept is summarized in Figure 3.

Figure 3: Illustration demonstrating the concept of balance between pro- and anti-remodelling adipokines in determining the resultant contribution to cardiac hypertrophy. Created with BioRender.com. Challenging as this may be, understanding their contributions and the underlying cellular and molecular mechanisms can provide the opportunity for the use of selective adipokine blockers for the treatment of heart failure particularly with heart failure associated with obesity. Moreover, the use of antiremodelling adipokines or their analogues offers the opportunity for additions to the currently available armamentarium for the treatment of heart failure.
Author Contributions
Conceptualization, MK; writing—original draft, MK, review and editing, MK, XTG. Both authors have read and approved the publication of this manuscript.
Funding:
Work cited from the authors’ laboratory was supported by the Canadian Institutes for Health Research (Grant # MOP 62764). MK was a recipient of a Tier I Canada Research Chair in Experimental Cardiology during the course of these studies.
Institutional Review Board Statement:
Not applicable
Informed Consent Statement:
Not applicable.
Data Availability Statement:
Not applicable.
Conflicts Of Interest:
The authors declare no conflicts of interests.
References
- Kyle, T.K.; Dhurandhar, E.J.; Allison, D.B. Regarding obesity as a disease: Evolving policies and their implications. Endocrinol. Metab. Clin. North Am. 2016, 45, 511-520. doi: 10.1016/j.ecl.2016.04.004.
View at Publisher | View at Google Scholar - Lingvay,I.; Cohen, R.V.; Roux, C.W.L.; Sumithran, P. Obesity in adults. Lancet 2024, 404, 972-987.doi: 10.1016/S0140-6736(24)01210-8.
View at Publisher | View at Google Scholar - Kenchaiah, S.; Evans, J.C.; Levy, D.; Wilson, P.W.; Benjamin, E,J.; Larson, M.G.; Kannel, W.B.; Vasan, R.S. Obesity and the risk of heart failure. N Engl J Med. 2002, 347, 305-13. doi: 10.1056/NEJMoa020245.
View at Publisher | View at Google Scholar - Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland,I.J.; Sanders, P.; St-Onge, M.P.; American Heart Association Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Epidemiology and Prevention; and Stroke Council. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984-e1010. doi: 10.1161/CIR.0000000000000973.
View at Publisher | View at Google Scholar - Koskinas, K.C.; Van Craenenbroeck, E.M.; Antoniades, C.; Blüher, M.; Gorter, T.M.; Hanssen, H.; Marx, N.; McDonagh, T.A.; Mingrone, G.; Rosengren, A.; Prescott, E.B. Obesity and cardiovascular disease: an ESC clinical consensus statement. Eur. J. Prev. Cardiol. 2025, 32,:184-220. doi: 10.1093/eurjpc/zwae279.
View at Publisher | View at Google Scholar - Mahabadi, A.A.; Massaro, J.M.; Rosito, G.A.; Levy, D.; Murabito, J.M.; Wolf, P.A.; O'Donnell, C.J.; Fox C,S.; Hoffmann, U. Association of pericardial fat, intrathoracic fat, and visceral abdominal fat with cardiovascular disease burden: the Framingham Heart Study. Eur. Heart J. 2009, 30, 850-856. doi: 10.1093/eurheartj/ehn573.
View at Publisher | View at Google Scholar - Upadhaya, S.; Le Jemtel, T.H. Epicardial Adipose Tissue and Heart Failure. Trends Cardiovasc. Med. 2025,35,339-340. doi: 10.1016/j.tcm.2025.02.008.
View at Publisher | View at Google Scholar - Janssen-Telders, C.; Eringa, E.C.; de Groot, J.R.; de Man, F.S.; Handoko, M. The role of epicardial adipose tissue remodelling in heart failure with preserved ejection fraction. Cardiovasc. Res. 2025, 121, 860-870. doi: 10.1093/cvr/cvaf056.
View at Publisher | View at Google Scholar - Whitman, J.; Kozaily, E.; Michos, E.D.; Silverman, D.N.; Fudim, M.; Mentz, R.J.; Tedford, R.J.; Rao V.N. Epicardial fat in heart failure and preserved ejection fraction: Novel insights and future perspectives. Curr. Heart Fail. Rep. 2025,22,13. doi: 10.1007/s11897-025-00700-5.
View at Publisher | View at Google Scholar - Ebong, I.A.; Goff, D.C. Jr.; Rodriguez, C.J.; Chen, H.; Bertoni, A.G. Mechanisms of heart failure in obesity. Obes. Res. Clin. Pract. 2014, 8, e540-8. doi: 10.1016/j.orcp.2013.12.005.
View at Publisher | View at Google Scholar - Alansari, H.; Lazzara, G.; Taha, M.B.; Gorthi, J.R. The impact of obesity on cardiovascular diseases: Heart failure. Methodist Debakey Cardiovasc. J. 2025, 21, 44-52. doi: 10.14797/mdcvj.1511
View at Publisher | View at Google Scholar - Piché, M.E.; Tchernof, A.; Després, J.P. Obesity phenotypes, diabetes, and cardiovascular diseases. Circ. Res. 2020, 126, 1477-1500. doi: 10.1161/CIRCRESAHA.120.316101
View at Publisher | View at Google Scholar - Cesaro, A.; De Michele, G.; Fimiani, F.; Acerbo, V.; Scherillo, G.; Signore, G.; Rotolo, F.P.; Scialla, F.; Raucci, G.; Panico, D.; Gragnano, F.; Moscarella, E.; Scudiero, O.; Mennitti, C.; Calabrò, P. Visceral adipose tissue Preprints.org (www.preprints.org) | NOT PEER-REVIEWED | Posted: 27 November 2025 doi:10.20944/preprints202511.2058.v1 © 2025 by the author(s). Distributed under a Creative Commons CC BY license. 23 of 38 and residual cardiovascular risk: a pathological link and new therapeutic options. Front. Cardiovasc. Med. 2023, 10, 1187735. doi: 10.3389/fcvm.2023.1187735.
View at Publisher | View at Google Scholar - Amundson, D.E.; Djurkovic, S.; Matwiyoff, G.N. The obesity paradox. Crit. Care Clin. 2010, 26, 583-596. doi: 10.1016/j.ccc.2010.06.004.
View at Publisher | View at Google Scholar - Dramé, M.; Godaert, L. The obesity paradox and mortality in older adults: A systematic review. Nutrients 2023, 15,1780. doi: 10.3390/nu15071780.
View at Publisher | View at Google Scholar - Horwich, T.B.; Fonarow, G.C.; Clark, A.L. Obesity and the obesity paradox in heart failure. Prog. Cardiovasc. Dis. 2018, 61,151-156. doi: 10.1016/j.pcad.2018.05.005.
View at Publisher | View at Google Scholar - Antonopoulos, A.S.; Tousoulis, D. The molecular mechanisms of obesity paradox. Cardiovasc. Res. 2017, 113, 1074-1086. doi: 10.1093/cvr/cvx106.
View at Publisher | View at Google Scholar - Pan, J.; Yin, J.; Gan, L.; Xue, J. Two-sided roles of adipose tissue: Rethinking the obesity paradox in various human diseases from a new perspective. Obes. Rev. 2023, 24, e13521. doi: 10.1111/obr.13521.
View at Publisher | View at Google Scholar - Pan, J.; Yin, J.; Gan, L.; Xue, J. Two-sided roles of adipose tissue: Rethinking the obesity paradox in various human diseases from a new perspective. Obes. Rev. 2023, 24, e13521. doi: 10.1111/obr.13521.
View at Publisher | View at Google Scholar - Butt, J.H.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæ k, L.; Gustafsson, F.; Kristensen, S.L.; Bruun, N.E.; Eiskjæ r, H.; Brandes, A.; Hassager, C.; Svendsen, J.H.; Høfsten, D.E.; Torp-Pedersen, C.; Schou, M.; Pehrson, S.; Packer, M.; McMurray, J.J.V.; Køber, L. Anthropometric measures and long-term mortality in nonischaemic heart failure with reduced ejection fraction: Questioning the obesity paradox. Eur. J. Heart Fail. 2025, 27, 527-536. doi: 10.1002/ejhf.3424. 20. Wertheimer, E.; Shapiro, B. The physiology of adipose tissue. Physiol. Rev. 1948, 28, 451-464. doi: 10.1152/physrev.1948.28.4.451
View at Publisher | View at Google Scholar - Kershaw, E.E.; Flier, J,S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548- 2556. doi: 10.1210/jc.2004-0395.
View at Publisher | View at Google Scholar - Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: an endocrine organ. Arch. Med. Sci. 2013, 9, 191-200. doi: 10.5114/aoms.2013.33181.
View at Publisher | View at Google Scholar - Scheja, L,; Heeren J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507-524. doi: 10.1038/s41574-019-0230-6.
View at Publisher | View at Google Scholar - Chaldakov, G.N.; Stankulov, I.S.; Hristova, M.; Ghenev, P. Adipobiology of disease: adipokines and adipokine-targeted pharmacology. Curr. Pharm. Des. 2003, 9, doi: 10.2174/1381612033455152.
View at Publisher | View at Google Scholar - Yang, F.T.; Stanford, K.I. Batokines: Mediators of Inter-Tissue Communication (a Mini-Review). Curr. Obes. Rep. 2022, 11,1-9. doi: 10.1007/s13679-021-00465-7.
View at Publisher | View at Google Scholar - Bays, H.E.; Kirkpatrick, C.F.; Maki, K.C.; Toth, P.P.; Morgan, R.T.; Tondt, J.; Christensen, S.M.; Dixon, D.L.; Jacobson, T.A. Obesity, dyslipidemia, and cardiovascular disease: A joint expert review from the Obesity Medicine Association and the National Lipid Association 2024. J. Clin. Lipidol. 2024, 18, e320-e350. doi: 10.1016/j.jacl.2024.04.001.
View at Publisher | View at Google Scholar - Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79-96. doi: 10.1161/CIRCRESAHA.115.302922.
View at Publisher | View at Google Scholar - Rao, V.N.; Zhao, D.; Allison, M.A.; Guallar, E.; Sharma, K.; Criqui, M.H.; Cushman, M.; Blumenthal, R.S.; Michos, E.D. Adiposity and Incident Heart Failure and its Subtypes: MESA (Multi-Ethnic Study of Atherosclerosis). JACC Heart Fail. 2018, 6, 999-1007. doi: 10.1016/j.jchf.2018.07.009.
View at Publisher | View at Google Scholar - Neeland, I.J.; Gupta, S.; Ayers, C.R.; Turer, A.T.; Rame, J.E.; Das, S.R.; Berry, .JD.; Khera, A.; McGuire, D.K.; Vega, G.L.; Grundy, S.M.; de Lemos, J.A.; Drazner, M.H. Relation of regional fat distribution to left ventricular structure and function. Circ. Cardiovasc. Imaging. 2013, 6, 800-807. doi: 10.1161/CIRCIMAGING.113.000532.
View at Publisher | View at Google Scholar - Raggi, P.; Stillman, A.E. Clinical Role of Epicardial Adipose Tissue. Can. J. Cardiol. 2025, Feb 17, S0828- 282X(25)00131-X. doi: 10.1016/j.cjca.2025.02.021
View at Publisher | View at Google Scholar - Vincenzi, M.; Nebigil, C.G. Uncovering the role of prokineticin pathway on Epicardial Adipose Tissue (EAT) development and EAT-associated cardiomyopathy. Trends Cardiovasc. Med. 2025, 35, 328-338. doi: 10.1016/j.tcm.2025.02.006.
View at Publisher | View at Google Scholar
