

Research Article | DOI: https://doi.org/10.31579/2834-8761/026
A Syndrome with Diabetes Mellitus: An Unusual Case
- Saccomanni Bernardino *
Orthopaedic and Traumatologic Surgery, ASL BARI viale Regina Margherita, 74 Altamura (BA), Italy.
*Corresponding Author: Saccomanni Bernardino, Orthopaedic and Traumatologic Surgery, ASL BARI, vial Regina Margherita, 74 Altamura (BA), Italy.
Citation: Sacco Manni Bernardino, Bernardino (2023), A Syndrome With Diabetes Mellitus: An Unusual Case, Clinical Endocrinology and Metabolism 2(4) DOI:10.31579/2834-8761/026
Copyright: © 2023, Saccomanni Bernardino. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 07 July 2023 | Accepted: 20 July 2023 | Published: 24 July 2023
Keywords: diabetes; congenital; syndrome
Abstract
Berardinelli-Seip syndrome is a congenital disorder of metabolism. It is characterized by insulin-resistant diabetes mellitus and deficiency of adipose tissue. The clinical and laboratory features of the syndrome are mostly due to fat deficiency, diabetes, or to manifestations of secondary hyperinsulinemia. Muscles appear hypertrophic, with flat feet. The combination of clinical and laboratory findings is characteristic and readily recognized once the components of the syndrome are known. WE report a case of a 3-year-old male, with a history of disease, who demonstrated many of the typical clinical and laboratory features of BSCL2.
Introduction
Berardinelli-Seip syndrome is a disorder of metabolism that results in lipodystrophy, endocrine abnormalities, and characteristic skeletal changes. It was originally described in Brazil by Berardinelli in 1954 [1] and subsequently confirmed in Norway by Seip in 1958 [2]. Since the initial description, 60 cohorts have been reported. The prevalence appears to range from 1 in 200,000 (Portugal) to 1 in 12,000,000 (USA) [3]. Berardinelli-Seip congenital lipodystrophy is also known as congenital generalized lipodystrophy, Seip syndrome, and lipoatrophic diabetes. The entity named lipodystrophy muscular hypertrophy [14] may also represent the same condition. Muscles appear hypertrophic. In addition, characteristic, but idiopathic, periarticular lytic lesions may be seen in some individuals. The combination of imaging, clinical, and findings is characteristic and readily recognized once the components of syndrome are known. We report a case of a 3-year-old male, with a history of disease, who demonstrated many of the typical clinical and laboratory features of BSCL2.
Materials And Methods
A 3-year-old male, came to our outpatient department, with the presenting complaints of abdominal distension, abnormal facies, darkening of the skin and excessive hair all over the body since early infancy (Fig. 1).
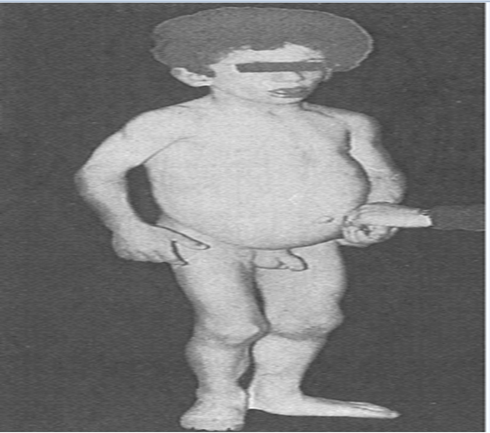
Figure 1: A 3-year-old male, with the presenting complaints of abdominal distension, mental retardation, abnormal facies, darkening of the skin and excessive hair all over the body since early infancy.
The child had a height of 96 cm and a weight of 15 Kg. He has lack of subcutaneous fat in all body parts, apparent muscular hypertrophy, coarse and hyperpigmented skin, acanthosis nigricans in the nape of the neck, with flat feet, excessive body hairs, hypertrophic genitalia and large superficial veins. He had hepatomegaly and splenomegaly without features of cirrhosis or hypertension. Mental retardation was present in child (Fig. 1). Laboratory investigations revealed normal hemogram. Liver enzymes were elevated (SGOT=80 IU/l, SGPT=143 IU/l, ALP=1717 IU/L); serum triglycerides were elevated (280 mg/dL); serum total cholesterol was normal. Blood sugar was normal. Plasma insulin levels were elevated, suggesting insulin resistance (fasting= 30 au/mL and postprandial = 38 au/mL).
X-ray of skull, trunk and limbs SHOWED normal bones. Echocardiography was normal.
In view of the typical dysmorphology, age of onset and hypertriglyceridemia, the diagnosis of THIS SYNDROME was kept.
The child is coming for follow-up since the last one-year. As per recommendations the child WAS put-on low-fat diet. The child has developed overt diabetes and serum triglycerides has somewhat decreased with dietary restriction. Special education of the child WAS advised because of the associated mental retardation.
Results And Discussion
Berardinelli-Seip congenital lipodystrophy is a rare autosomal recessive disorder. It has been variously described as generalized lipodystrophy, congenital lipodystrophy, Seip-Lawrence syndrome and lipoatrophic diabetes (in USA).
BSCL families are classified into BSCL1, BSCL2 and BSCLX [8]. BSCL1, prevalent in African-American population, is the milder variety presenting in the second or third decade of life. Garg A, Wilson R, Barnes R, Arioli E, Zaidi Z, Gerakan F [4] first identified the gene for BSCl1 on chromosome 9q34. BSCL2 is more severe with onset in the neonatal period or early infancy. Most have mental retardation. It is prevalent in Portugal, Lebanon, Norway and Middle East. The locus for BSCl2, has been identified on chromosome 11q13 by Marge J, Delepine M, Khalaf E, Genne-Dahl T Jr, Van Maldegem L, Sobel E [6]; the prevalence of BSCL2 is somewhat more than that of BSCL1. BSCLX families are very rare; they show evidence against co-segregation with either 9q34 or 11q13.Argwall AK, Arioli E, De Almeida S, Akko N, Taylor SI, Bowcock AM [7] have detected different mutations of the gene encoring 1-acylglycerol-3-phosphateO-acyltransferase 2 in 20 affected individuals showing linkage to chromosome 9q34 (BSCL1). The AGPA T2 enzyme is involved in the biosynthesis of triacylglycerol and glycerophospholipids.
In the 11q13 linked families (BSCL2) various homozygous and compound heterozygous mutations were found in a previously unidentified gene, termed seeping. More than 15 different allelic variations of the BSCL2 gene have been described [6, 8]. The seeping gene encodes for a 398-amino acid protein of unknown function. Marge J, Delepine M, Khalaf E, Genne-Dahl T Jr, Van Maldegem L, Sobel E [6] in 2001 indicated than this protein could be a transmembrane protein. Windlassing C, Auer-Grumbach M, Ibori J, Patel Hour, Petek E, Horl G [9] in 2004 showed that seeping is an integral membrane protein of the endoplasmic reticulum. This gene is widely expressed, with particularly high expression levels in brain and testis.
In all BSCL subjects, there is near total absence of adipose tissue.
Dietary and endogenous fat is aberrantly stored in metabolically important tissues like muscle and liver. This leads to severe insulin resistance and ultimately a difficult to control diabetes mellitus. Other metabolic consequences include hypertriglyceridemia leading to hepatic steatosis and ultimately to cirrhosis and death due to hepatic failure. For unknown reasons many subjects develop hypertrophic cardiomyopathy that can lead to death from cardiac failure [8]. Association with bone cyst has been reported; it was previously described as Brunzell syndrome [5].
Most of the clinical features of the syndrome can be explained as being due to either the consequences of the fat deficiency, diabetes, or to manifestations of secondary hyperinsulinemia, which results from the failure of the tissue to respond to insulin.
Hyperinsulinemia results in an elevated basal metabolic rate and excessive food intake. It has been recently recognized that the central nervous system can sense and respond to input from hormones such as insulin and leptin that are usually secreted in proportion to body energy stores [10]. Children with BSCL experience rapid increases in height, weight, muscle mass, and skeletal mineralization.
Growth may confer some protection against the hypermetabolic effects as metabolic disorders tend to become more severe after puberty. Children with BSCL may also demonstrate advanced bone age on radiographs. Hands and feet are large, as in acromegaly, although growth hormone levels are normal. Patients with BSCL also frequently suffer from acanthosis nigricans, a hyperpigmentation disorder that is generally thought to be caused by elevated levels of insulin or insulin-like growth factors.
In absence of adequate reservoirs of adipose storage, dietary and endogenous fat is aberrantly stored in other tissues such as muscle and liver [3]. Therefore, although it may see counter-intuitive given the general absence of fat, hepatomegaly and muscle enlargement may result from fatty infiltration. A recent work indicates that insulin resistance is associated with increased levels of lipid within skeletal muscle cells [11]. It is thought that these increased levels result in impaired levels result in impaired energy metabolism and may play a central role in the pathogenesis of insulin resistance. Morphological and functional of the skeletal musculature of patients with this disease suggests that increased muscle mass should more properly be termed hyperplasia rather than hypertrophy, since patients with BSCL do not demonstrate increased strength. Musculoskeletal symptoms in BSCL are probably due to the combined effects of muscle hypertrophy and impaired energy metabolism.
Laboratory findings of BSCL are due to diabetes and to abnormalities of hepatic, renal, and pancreatic function.
Despite hepatomegaly, in most patients the liver function is well preserved. However, hepatic dysfunction may cause triglyceride and glycogen levels to increase and severe cases of cirrhosis have been reported. Decreased renal function is due to diabetes mellitus. Proteinuria is often present. As in patients with the common insulin resistance syndrome, individuals with BSCL are subject to an increased risk of atherosclerosis [12]. Hyperglycemia due to diabetes mellitus and low HDLC may be observed as early as in infancy.
Leptin is a peptide hormone that is secreted by fat cells. It has many physiological effects, one of which is oxidation of fatty acids, reducing the lipid content of cells. It has been shown that leptin therapy may help to reverse the effects of insulin resistance. Unfortunately, the usefulness of leptin is limited by side effects and the rapid occurrence of tachyphylaxis [13].
Conclusion
In our case, we suspected generalized lipoatrophy. The diagnosis was confirmed after the typical dysmorphology was noted and hypertriglyceridemia and hyperinsulinemia were detected. In conclusion, we have presented a patient with features of classic type 2 BSCL. The condition can be recognized by a characteristic combination of clinical and laboratory features, and sheds light on some of the physiological functions of adipose tissue.
Acknowledgment:
NONE
References
- Berardinelli W. (1954). An undiagnosed endocrinometabolic syndrome: report of two cases [abstract], J Clin Endor, 14: 193-194.
View at Publisher | View at Google Scholar - Seip M. (1959). Lipodystrophy and gigantism with associated endocrine manifestation: a new diencephalic syndrome? [abstract], Acta Pediatric, 48: 555-574.
View at Publisher | View at Google Scholar - Mandal K, Aneja S, Khan A. (2006). Berardinelli-Seip congenital lipodystrophy [abstract]. Indian Pediatric, 43: 440-445.
View at Publisher | View at Google Scholar - Garg A, Wilson R, Barnes R, Arioli E, Zaidi Z, et all., (1999). A gene for congenital generalized lipodystrophy maps to human chromosome 9q34, J Clin Endor Meta, 84: 3390-3394.
View at Publisher | View at Google Scholar - Brunzell JD, Shankle SW, Bethune JE. (1968). Congenital generalized lipodystrophy and systematic cystic angiomatosis: the simultaneous occurrence of two unusual syndromes in a single family [abstract], Ann Inter Med, 69: 501-516.
View at Publisher | View at Google Scholar - https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Magre+J%2C+Delepine+M%2C+Khalaf+E%2C+Genne-Dahl+T+Jr%2C+Van+Maldegem+L%2C+et+all.%2C+%282001%29.+Identification+of+the+gene+altered+in+Berardinelli-Seip+congenital+lipodystrophy+on+chromosome+11q13.%2C+Nature+Genet%2C+28%3A+365-370.&btnG=
View at Publisher | View at Google Scholar - Argwall AK, Arioli E, De Almeida S, Akko N, Taylor SI, et all., (2002). AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nature Genet ,31: 21-23.
View at Publisher | View at Google Scholar - Maldergem Van L, Marge J, Khalaf TE, Genne-Dahl Jr, Delepine M, et all., (2002). Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy, J Med Genet, 39: 722-733.
View at Publisher | View at Google Scholar - Windpassinger C, Auer-Grumbach M, Ibori J, Patel Hour, Petek E, et all., (2004). Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and silver syndrome. Nature Genet, 36: 271-276.
View at Publisher | View at Google Scholar - Schwartz MW, Porte D Jr. (2005). Diabetes, obesity, and the brain [abstract]. Science, 307: 375-379.
View at Publisher | View at Google Scholar - Torriano M. (2007). Measuring muscle lipids with 1H-MR spectroscopy. Skeletal Radiol, 36: 607-608.
View at Publisher | View at Google Scholar - Hegele R. (2001). Atherosclerosis associated with monogenic insulin resistance [abstract]. Circulation, 2225-2229.
View at Publisher | View at Google Scholar - Oral EA, Simha V, (2002). Leptin-replacement therapy for lipodystrophy [abstract]. New England J Med, 346: 570-578.
View at Publisher | View at Google Scholar - Senior B. (1961). Lipodystrophy muscular hypertrophy [abstract], Arch Dis Child, 36: 426-431.
View at Publisher | View at Google Scholar
