

Research Article | DOI: https://doi.org/10.31579/2834-8486/017
SuccinatSuccinate Dehydrogenase (Sdh) Deficient Renal Cell Carcinoma: Review and Updatee DehydrogenasSuccinate Dehydrogenase (Sdh) Deficient Renal Cell Carcinoma: Review and Updatee (Sdh) Deficient Renal Cell Carcinoma: Review and Update
- Anthony Kodzo-Grey Venyo *
North Manchester General Hospital, Department of Urology, Delaunays Road, Crumpsall. M8 5RB. United Kingdom.
*Corresponding Author: Anthony Kodzo-Grey Venyo, North Manchester General Hospital, Department of Urology, Delaunays Road, Crumpsall. M8 5RB. United Kingdom.
Citation: Anthony K Venyo., and Ebenezer O Ajiboye., (2024). Succinate Dehydrogenase (SDH) Deficient Renal Cell Carcinoma: Review and Update. International Journal of Clinical Therapeutics. 3(4); DOI:10.31579/2834-8486/017
Copyright: © 2024, Anthony Kodzo-Grey Venyo. Nguyen, this is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 08 July 2024 | Accepted: 29 July 2024 | Published: 28 August 2024
Keywords: renal cell carcinoma; rcc; succinate dehydrogenase; sdh; sdh-deficient rcc; incidence; diagnosis, pathology, high mitotic index; necrosis; rare
Abstract
Succinate dehydrogenase (SDH) is a mitochondrial enzyme complex which is comprised of 4 protein subunits (SDHA, SDHB, SDHC, and SDHD). Germ line mutations of the genes encoding these SDH subunits emanate in hereditary syndromes harbouring pheochromocytomas/paragangliomas, gastrointestinal stromal tumours, renal cell carcinomas, and pituitary adenomas. SDH-deficient renal cell carcinomas are uncommon, with a mean age at initial manifestation of 38 years to 40 years. Upon histopathology examination, these neoplasms do demonstrate a typifying appearance which includes a solid, nested, or tubular architecture with variable cysts. The cells in SDH-deficient renal cell carcinomas tend to be typically cuboidal, they have indistinct cell borders and eosinophilic cytoplasm, as well as they demonstrate flocculent intracytoplasmic inclusions. Loss of immunohistochemical staining for SDHB is the hallmark of these tumours. Even though majority of SDH-deficient renal cell carcinomas have tended to be indolent clinically, some of the tumours may portend an aggressive biological behaviour, especially tumours that contain a high nuclear grade, tumour necrosis, or sarcomatoid differentiation. Accurate classification of these tumours is pertinent and pivotal for clinical follow-up, screening, and genetic evaluation of the patients and other family members for this hereditary tumour syndrome.
Succinate dehydrogenase (SDH) is the key enzyme which converts succinate to fumarate in the tricarboxylic acid cycle, and SDH comprises of 4 subunits: SDHA, SDHB, SDHC, and SDHD. SDHA and SDHB are catalytic domains, whereas SDHC and SDHD are stated to be ubiquinone-binding and membrane-anchorage domains. Germ line mutations within the gene encoding SDH do predispose patients to neoplastic transformation when there is a loss of the remaining wild-type allele in the somatic cells for example loss of heterozygosity, which does cause a complete loss of enzyme function, which emanates in the intracytoplasmic accumulation of succinate. This is ensued by the metabolic reprograming of the “tumour microenvironment” despite normal oxygen levels, and does provide an advantageous environment for survival of the tumour, and does emanate in a series of tumour syndromes that are referred to as hereditary paraganglioma-pheochromocytoma syndromes.
SDHD and SDHC mutations are typically associated with multifocal head and neck paragangliomas and less frequently with adrenal pheochromocytomas and extra-adrenal paragangliomas, which are usually benign, with rare cases of metastasis. SDHB mutations mainly emanates in predisposition to extra-adrenal paragangliomas with high malignant potential, and, to a lesser extent, adrenal pheochromocytomas and head and neck paragangliomas. Furthermore, malignant paragangliomas, SDHB mutations also tend to be associated with malignant tumours of the extraparaganglial system, including renal cell carcinoma (RCC) and thyroid carcinoma. In addition to multiple paragangliomas, SDHA mutations have also been demonstrated to be associated with neurodegenerative diseases, for example, an early-onset encephalopathy which is referred to as Leigh syndrome, and a late-onset optic atrophy, ataxia, and myopathy. Mutations of all of the above subunits had been demonstrated to be associated with gastrointestinal stromal tumours (GISTs) with wild-type c-Kit or platelet-derived growth factor receptor α.
SDH-deficient RCC was first identified in 2004 and had been accepted by the 2016 World Health Organization classification of renal tumours as a unique subtype of RCC. In view of their strong syndromic association and distinct natural history, it is pivotal to appropriately identify as well as classify these tumours. Recent large studies of this tumour had demonstrated a wide spectrum of morphology and genetic alterations within the tumour.
Some summations related to SDH-deficient RCCs include the ensuing:
- SDH-deficient RCCs are uncommon and had been estimated to account for 0.05% to 0.2% of all RCCs.
- The age at initial presentation and diagnosis have ranged from 14 years to 76 years, with a mean age of 38 years to 40 years and a slight male predominance.
- Most patients who are afflicted by SDH-deficient RCCs do have germ line mutations in SDHB, SDHC, SDHA, or SDHD. These mutations emanate in hereditary syndromes harbouring pheochromocytomas/paragangliomas, GISTs, RCCs, and pituitary adenomas.
- SDH-deficient GISTs and paragangliomas could also occur within the syndromic, nonhereditary Carney triad of paraganglioma, pulmonary chondroma, and SDH-deficient GIST, which is caused by SDHC promoter-specific CpG island hypermethylation; nevertheless, this epimutation had been recently stated not to have been reported so far in SDH-deficient RCCs
- The lifetime risk of renal tumours in patients who have been afflicted with the SDHB gene mutation had been estimated to be 14%.
- It has been recommended that in view of this lifelong risk, genetic testing in the appropriate clinical context should be considered for patients who this constellation of tumours.
- Clinically, majority of SDH-deficient RCCs do manifest as small, organ-confined tumours that tend to diagnosed incidentally by means of radiology imaging for something else.
- Rarely, these tumours may manifest with metastatic disease.
- Multifocal or bilateral tumours had been identified found in as many as 30% of patients at long-term follow-up
- Some of the take home messages include:
- Clinicians and Urologists globally need to be aware that SDH-deficient renal cell carcinomas do exist and they should have a high-index of suspicion for these tumours based upon the pathology examination features of the tumours, family history of similar types of tumours and clinicians should also be aware of factors of poor prognostication including high mitotic index, necrosis in the tumours and other factors.
- Clinicians should continue to report cases of SDH-deficient RCCs they treat so that more lessons would be learnt globally about the biological behaviour of these rare renal tumours.
Introduction:
It has been pointed out that cancer of the Kidney is fundamentally a metabolic disease and that each of the genes that are known to cause kidney cancer including the ensuing: VHL, MET, FLCN, fumarate hydratase, succinate [1] dehydrogenase, TSC1, TSC2, TFE3, TFEB, MITF, as well as PTEN is involved in the fundamental cellular processes that regulate the cell’s response to sensing oxygen, iron, nutrients and energy. [1] [2] It has been iterated that understanding of the fundamental metabolic basis of kidney cancer should be able to provide the foundation for the development of novel approaches to the treatment of this disease. [1]
It has been documented that Germline mutation of the Krebs cycle (tricarboxylic acid cycle) enzyme, fumarate hydratase, has been noted to be associated with an aggressive form of type II papillary kidney cancer in patients who are afflicted with Hereditary Leiomyomatosis as well as Renal Cell Carcinoma (HLRCC). [1] [3] [4] [5] It has also been pointed out that with regard to fumarate hydratase-deficient kidney cancer, a prototypic example of the Warburg effect in cancer, oxidative phosphorylation tends to be severely compromised as well as the cells do undergo a metabolic shift to aerobic glycolysis to generate ATP and other metabolites that are necessitated for rapid growth and cell division. [1] [6] [7] [8] [9] It has furthermore been pointed out that the emanation or result is an extremely aggressive, lethal form of cancer of the kidney that is associated with a very high metabolic rate which does have a propensity to metastasize when the primary tumour is noted to be very small and measuring less than 1 centimetre at the onset. [1] [4]
It has been iterated that a second form of inherited carcinoma of kidney that is typified by a Krebs cycle gene mutation had been initially reported by Vanharanta, and associates [10], who had reported three patients who had cancer of the kidney in families that had a germline mutation of succinate dehydrogenase B (SDHB) that was also associated with hereditary paraganglioma (PGL), a group of diseases that had been known to be associated with head and neck PGL which can include a history of pheochromocytoma (PCC).[1] [10] It has furthermore been pointed out that whilst long noted as having a hereditary component, the genes that are associated with this condition were difficult to identify as mapping studies had linked suspected genes to different types of chromosomal regions. Over the preceding decade which was about two decades from the time of publication of this article, the genetic basis of this syndrome had been identified and four specific syndromes had been linked to gene mutations in a multi-complex Krebs cycle/electron transport chain enzyme, succinate dehydrogenase (SDH). [1] [11] [12] [13] [14] This enzyme is composed of four subunits (SDHA, SDHB, SDHC, and SDHD) and catalyses the oxidation of succinate to fumarate. Furthermore, germline mutations of SDHB, SDHC and SDHD had been identified in patients who have a combination of gastrointestinal stromal tumours (GISTs) and paraganglioma (PGL) and patients who had manifested solely with GIST. [1] [15] [16] Multiple reports had documented the identification of the presence of Renal Cell Carcinoma (RCC) in patients who had SDHB mutations either with or without a personal or family history of PGL and/or PCC. [10] [17] [18] [19] [20] [21] [22] [23] In addition, a recent investigation had described 2 individuals who had Cowden or Cowden-like syndromic features which included: Renal Cell Carcinoma (RCC); however, no PTEN mutation, had reported SDHD gene variants which had affected the AKT and/or MAPK pathways. [1] [24] Pursuant to the submission of this work for publication, Malinoc and associates [25] had reported an individual patient who had a germline SDHC gene mutation with personal and family history of PGL who was then found to have Renal Cell Carcinoma (RCC) tumours associated with loss of the wild-type SDHC allele. [1] [25]. Considering that not much is known generally by all clinicians globally about the clinical manifestations and management as well as outcome pursuant to the treatment of patients who have SDH mutation-associated Renal Cell Carcinoma, (SDH-RCC), it is important to review and update the literature. The ensuing article contains a review and update of the literature that has been focussed upon Succinate Dehydrogenase (SDH) Deficient Renal Cell Carcinoma, which has been divided into two parts: (A) Overview which contains miscellaneous discussions to various general aspects of Succinate Dehydrogenase (SDH) Deficient Renal Cell Carcinoma and (B) Miscellaneous Narrations and Discussions from Some Case Reports, Case Series and Studies Related to Succinate Dehydrogenase (SDH) Deficient Renal Cell Carcinoma:
Aim: To review and update the literature on Succinate Dehydrogenase (SDH) Deficient Renal Cell Carcinoma.
Methods
Various internet data bases were searched including: Google: Google Scholar; Yahoo; and PUBMED. The search words that were used included: Succinate Dehydrogenase Deficient Renal Cell Carcinoma: SDH deficient renal cell carcinoma; Succinate Dehydrogenase Deficient Renal Cell Cancer: SDH deficient kidney cancer; SDH deficient carcinoma of kidney; Succinate Dehydrogenase (SDH) Deficient Carcinoma of kidney. Sixty (60) references were identified which were used to write the review and update article on Succinate Dehydrogenase (SDH) Deficient Renal Cell Carcinoma, which has been divided into two parts: (A) Overview which contains miscellaneous discussions to various general aspects of Succinate Dehydrogenase (SDH) Deficient Renal Cell Carcinoma and (B) Miscellaneous Narrations and Discussions from Some Case Reports, Case Series and Studies Related to Succinate Dehydrogenase (SDH) Deficient Renal Cell Carcinoma:
Results:
[A} Overview
Definition / general statement [26]
•It has been iterated that Succinate dehydrogenase (SDH) deficient renal cell carcinoma had been defined by the World Health Organization (WHO) as a malignant epithelial tumour which is composed of vacuolated eosinophilic to clear cells with loss of immunohistochemical expression of SDHB, which a marker of dysfunction of mitochondrial complex II (WHO 2016) [26]
Essential features
The essential features of SDH had been summated as follows: [26]
•In SDH, microscopy examination of the kidney tumour does demonstrate characteristic flocculent cytoplasmic vacuoles that contains a pale eosinophilic, wispy or bubbly appearance and low-grade nuclei (at least focally)
•SDH might have high grade areas
•In SDH, oncogenesis tends to be driven by metabolic derangements due to double hit inactivation of SDH genes, leading to dysfunction of mitochondrial complex II
•Majority of patients who have SDH do have a germline mutation in an SDH gene, usually SDHB
Terminology [26]
•It has been pointed out that terminology which is sometimes utilised but not recommended is SDHB negative renal carcinoma or SDH deficient renal oncocytoma [26]
Epidemiology
•It has been iterated that SDH is uncommon. [26]
•It has been iterated that SDH does account for 0.05% to 0.2% of all renal cell carcinomas (WHO 2016) [26]
•It has been documented that the median age of patients who have SDH is 35 years and that the age range of patients who have SDH has varied between 14 years to 76 years as well as that the male to female (M/F); ratio of patients who had been diagnosed as having SDH is 1:8:1 [26]
•It has also been documented that SDH had been stated to be bilateral in 26% of cases of SDH of kidney. [27]
Sites
•It has been iterated that SDH does afflict the kidney [26]
Pathophysiology [26]
•It has been iterated that majority of cases of SDH, do occur in setting of germline mutation of an SDH gene and that the neoplasia occurs with double hit inactivation, leading to dysfunction of mitochondrial complex II, increased reactive oxygen species, DNA damage and HIF1α stabilization [28]
•It has been documented that SDHB has tended to be most commonly affected in SDH deficient RCC, then SDHC. [26]
•It has been iterated that with regard to SDH, the tumour could be rarely SDHA or SDHD, which is different for other SDH deficient tumours [26]
Clinical features
•It has been iterated that SDH often has tended to be confined to the kidney at the time of presentation [26]
•It has been documented that SDH does manifests with flank pain or as incidental finding. [26]
•It has been iterated that in SDH, personal or family history of paragangliomas or SDH deficient gastrointestinal stromal tumour may be present (WHO 2016) [29]
Prognostic factors [26]
•It has been iterated that in SDH, the overall metastasis rate has been about 33%, based upon 27 reported cases; however, but the development of metastasis uncommon if the tumour upon pathology examination demonstrates only low-grade features [27]
•It has been iterated that SDH of kidney tends to associated with higher metastasis rate if pathology examination of the excised tumour demonstrates high grade nuclei (70% metastasize) or coagulative necrosis is found present in that 4 out of 4 cases had metastasized, [27]
Treatment
The treatment of SDH of the kidney has been summarized as follows: [26]
•It has been iterated that treatment of SDH of kidney does entail resection of the kidney tumour and that further treatment is dependent upon the pathology grade / stage of the tumour as well as the treatment may include targeted therapy [30]
•It has been advised that all patients who are diagnosed as having SDH deficient RCC should be offered genetic testing (WHO 2016) [26]
Macroscopy examination features.
The macroscopy pathology examination features of SDH of kidney had been summated as follows: [26]
•Gross examination of SDH of kidney does tend to demonstrate: Well circumscribed, solid with red / brown cut surface, variable multi-cystic change, generally no necrosis tends to be visualised upon microscopy examination of SDH. [26]
•Upon macroscopy examination of SDH tumour of the kidney, the tumour usually tends to be confined to kidney with no involvement of renal sinus, vein or fat. [26]
Microscopic (histologic) description
The microscopy pathology examination features of SDH of the kidney had been summated as follows: [26]
•Microscopy examination of specimens of SDH of kidney does demonstrate well- circumscribed or pushing border, commonly the tumour entraps the kidney tubules
•Microscopy histopathology examination of a kidney tumour containing SDH tends to demonstrate solid, nested or tubular growth pattern with scattered cysts containing eosinophilic material
•Microscopy histopathology examination of an SDH kidney tumour does demonstrate neoplastic cells that have smooth nuclear contours and fine chromatin with no nucleoli
•It has been iterated that in SDH of kidney, microscopy histopathology examination of the kidney tumour specimen does tend to demonstrate typifying finding which entails flocculent cytoplasmic vacuoles with a pale eosinophilic, wispy or bubbly appearance with low grade nuclei
O This morphology is stated to be commonly diffuse and should be at least focal (WHO 2016)
•SDH of kidney upon microscopy examination may be demonstrated to contain areas of higher-grade nuclei, necrosis, sarcomatoid change
•Variant morphologies had been reported in cases of SDH of kidney but these are rare in absence of more characteristic low-grade regions (WHO 2016)
Immunohistochemistry staining study findings in SDH of kidney. [26] (See table 1 for the immunohistochemistry staining pattern of various types of kidney tumours).
Positive stains [26]
It has been iterated that SDH tumour of kidney does exhibit positive immunohistochemistry staining for the following tumour markers: [26]
•PAX 8.
•Focal positive staining for pancytokeratin and
•CAM 5.2 and
•EMA [31]
Negative stains
It has been iterated that SDH tumour of kidney does exhibit negative immunohistochemistry staining for the following tumour markers: [26]
•In SDH of kidney there tends to be loss of SDHB immunohistochemical staining and it has been advised that pathologists should be cautious on overinterpretation of negativity in tumours with very clear cytoplasm)
O Loss of SDHB immunohistochemical staining indicates disruption of the mitochondrial complex 2 for any reason, not just SDHB gene mutation. [27]
•CK7, CAIX, RCC, c-kit (mast cells only), vimentin [31], neuroendocrine markers
•There tends to be minimal AMACR staining. [26]
Immunohistochemistry panels of various types of renal cell carcinoma can be seen at original site in reference [26]
Additional Supporting References for the information can be found in references that include [32] [33] [34] [35] [36] [37]:
[32] Zhao W, Tian B, Wu C, Peng Y, Wang H, Gu WL, Gao FH. DOG1, cyclin D1, CK7, CD117 and vimentin are useful immunohistochemical markers in distinguishing chromophobe renal cell carcinoma from clear cell renal cell carcinoma and renal oncocytoma. Pathol Res Pract. 2015 Apr;211(4):303-307. doi: 10.1016/j.prp.2014.12.014. Epub 2014 Dec 30. PMID: 25596994. https://pubmed.ncbi.nlm.nih.gov/25596994/
[33] Michalova K, Tretiakova M, Pivovarcikova K, Alaghehbandan R, Perez Montiel D, Ulamec M, Osunkoya A, Trpkov K, Yuan G, Grossmann P, Sperga M, Ferak I, Rogala J, Mareckova J, Pitra T, Kolar J, Michal M, Hes O. Expanding the morphologic spectrum of chromophobe renal cell carcinoma: A study of 8 cases with papillary architecture. Ann Diagn Pathol. 2020 Feb; 44:151448. doi: 10.1016/j.anndiagpath.2019.151448. Epub 2019 Dec 14. PMID: 31918172.
[34] Epstein JI, Egevad L, Humphrey PA, Montironi R; Members of the ISUP Immunohistochemistry in Diagnostic Urologic Pathology Group. Best practices recommendations in the application of immunohistochemistry in the prostate: report from the International Society of Urologic Pathology consensus conference. Am J Surg Pathol. 2014 Aug;38(8): e6-e19. doi: 10.1097/PAS.0000000000000238. PMID: 25029122. https://pubmed.ncbi.nlm.nih.gov/25029122/
[35] Iczkowski KA, Czaja RC. Eosinophilic Kidney Tumors: Old and New. Arch Pathol Lab Med. 2019 Dec;143(12):1455-1463. doi: 10.5858/arpa.2019-0203-RA. Epub 2019 Aug 12. PMID: 31403331. https://pubmed.ncbi.nlm.nih.gov/31403331/
[36] Ng KL, Ellis RJ, Samaratunga H, Morais C, Gobe GC, Wood ST. Utility of cytokeratin 7, S100A1 and caveolin-1 as immunohistochemical biomarkers to differentiate chromophobe renal cell carcinoma from renal oncocytoma. Transl Androl Urol. 2019 May;8(Suppl 2):S123-S137. doi: 10.21037/tau.2018.11.02. PMID: 31236330; PMCID: PMC6559932. https://pubmed.ncbi.nlm.nih.gov/31236330/
[37] Liu YJ, Ussakli C, Antic T, Liu Y, Wu Y, True L, Tretiakova MS. Sporadic oncocytic tumors with features intermediate between oncocytoma and chromophobe renal cell carcinoma: comprehensive clinicopathological and genomic profiling. Hum Pathol. 2020 Oct;104:18-29. doi: 10.1016/j.humpath.2020.07.003. Epub 2020 Jul 13. PMID: 32673684. https://pubmed.ncbi.nlm.nih.gov/32673684/
Molecular / cytogenetics description
The molecular / cytogenetics features of SDH had been summated as follows: [26]
•In cases of SDH of kidney, there tends to be: No mutations in VHL, PIK3CA, AKT, mTOR, MET or TP53 genes (WHO 2016) [26]
•Genes for succinate dehydrogenase subunits (SDHA, SDHB, SDHC, SDHD) do encode proteins which are part of mitochondrial complex II, which links the Krebs cycle and electron transport chain [26] [23]
Differential diagnosis
Some of the differential diagnoses of SDH had been summated as follows: [26]
•Eosinophilic variant of chromophobe RCC:
oThese tumours tend to have: Prominent cell borders and crinkled / raisinoid nuclei with binucleation
oAlso, CK7+ with no loss of SDHB expression
•Eosinophilic variant of clear cell Renal Cell Carcinoma (RCC): CAIX+, vimentin+
•Oncocytoma: often has myxohyaline stroma; CKIT+ with no loss of SDHB expression [31]
•Papillary renal cell carcinoma (RCC) (type 2):
O Cytoplasm is eosinophilic but not as pale wispy/bubbly
O Nuclei often pseudostratified
•Loss of SDHB immunohistochemical expression is rare in other carcinomas [38] [B] Miscellaneous Narrations and Discussions From Some Case Reports, Case Series and Studies Related to Succinate dehydrogenase kidney cancer SDH of the Kidney.
In 2012, Ricketts et al. [1] iterated that recently, a new renal cell cancer syndrome had been linked to germline mutation of multiple subunits (SDHB/C/D) of the Krebs cycle enzyme, succinate dehydrogenase. Ricketts et al. [1] reported their experience with the diagnosis, assessment as well as treatment of this new form of hereditary kidney cancer. Ricketts et al. [1] enrolled patients who had suspected hereditary kidney cancer upon a National Cancer Institute institutional review board approved protocol in order to study inherited forms of kidney cancer. Ricketts et al. [1] iterated that individuals from families with germline SDHB, SDHC and SDHD mutations, and kidney cancer had undergone comprehensive clinical and genetic evaluation. Ricketts et al.
[1] summated their results as follows:
•They had evaluated a total of 14 patients from 12 SDHB mutation families.
•Patients had manifested with renal cell cancer at an early age of 33 years, and the age range at their initial presentations had varied from 15 years to 62 years.
•Metastatic kidney cancer had developed in 4 and some families had no manifestation other than kidney tumours.
•An additional family with 6 individuals who were found to have clear cell renal cell cancer which had manifested at a young average age of 47 years, with a presentation age range of between 40 years and 53 years was identified with a germline SDHC mutation (R133X).
•Metastatic disease had developed in 2 of these family members.
• A patient who had a history of carotid body paragangliomas and an aggressive form of kidney cancer was assessed from a family with a germline SDHD mutation.
Ricketts et al. [1] made the ensuing conclusions:
•SDH mutation associated renal cell carcinoma could be an aggressive type of kidney cancer, particularly in younger individuals.
•Even though detection and management of early tumours most often tends to be associated with a good outcome, based upon their initial experience with these patients and their long-term experience with hereditary leiomyomatosis and renal cell carcinoma, they had recommended careful surveillance of patients at risk for SDH mutation associated renal cell carcinoma and wide surgical excision of renal tumours.
Gill et al. [39] stated the ensuing:
•Germline succinate dehydrogenase B (SDHB) mutation causes pheochromocytoma/paraganglioma syndrome type 4 (PGL4).
•PGL4 is typified by pheochromocytoma and paraganglioma, type 2 (SDHB negative) gastrointestinal stromal tumours and renal tumours, which usually tend to be classified as carcinoma.
Gill et al. [39] reported 4 kindreds with 5 PGL4-associated kidney tumours. Four of the tumours had occurred preceding the age of 30 years, 4 of the tumours were found in the left kidney, 3 of the tumours were in female patients, and 4 of the tumours had demonstrated consistent but previously unrecognized morphology. The tumours upon pathology examination, adjudge to be composed of cuboidal cells with bubbly eosinophilic cytoplasm and indistinct cell borders. Many of the cells were noted upon pathology examination to have displayed distinctive cytoplasmic inclusions, which were noted to be vacuolated or to have contained eosinophilic fluid-like material. The cells upon pathology examination were noted to be arranged in solid nests or in tubules encompassing central spaces. The tumours were also noted to be well-circumscribed or lobulated and frequently exhibited cystic change. Benign tubules or glomeruli of the kidney containing the tumour were often found to be entrapped at the edges of the tumours. The fifth tumour was noted to have lacked these features but had displayed sarcomatoid dedifferentiation. Immunohistochemistry staining studies of the tumour for SDHB was found to be completely negative in all 4 available tumours. Death from metastatic disease had occurred in the patient who had dedifferentiated tumour 1 year pursuant to the diagnosis, whereas the other 4 tumours had been cured by local excision alone, pursuant to a mean follow-up of 11 years and a follow-up range between 2 years and 30 years. Gill et al. [39] made the ensuing conclusions:
•Morphology supported by negative immunohistochemistry for SDHB could be utilised to identify kindreds with germline SDHB mutations (PGL4 syndrome) manifesting with this unique type of kidney tumour.
•These kidney tumours appeared to have a good prognosis pursuant to complete excision unless there is sarcomatoid dedifferentiation.
Gill et al. [23] in 2021, published ensuing educative iterations:
•The genes for the succinate dehydrogenase subunits A, B, C, and D (SDHA, SDHB, SDHC, and SDHD, respectively) do encode proteins which constitute part of the mitochondrial complex II, which does link the Krebs cycle and the electron-transport chain.
•Heterozygous germline mutations of SDHB, SDHC, as well as SDHD do cause the well-characterized familial pheochromocytoma-paraganglioma syndromes that ae known respectively as PGL4, PGL3, and PGL1. [40]
•SDHB, SDHC, and SDHD mutations had also been linked to gastrointestinal stromal tumour, and SDHB and SDHD had been linked to renal-cell carcinoma.
•These kindreds are rarely recognized when they do manifest with gastrointestinal stromal tumour and they tend virtually never to be recognized when they manifest with renal-cell carcinoma.
•This lack of recognition had been unfortunate, taking into consideration, the malignant potential of all three component tumours, especially, in PGL4 that is caused by SDHB mutation), which had been documented to be especially associated with malignant pheochromocytoma-paraganglioma and renal-cell carcinoma [41]
•When any component of the mitochondrial complex II is lost, evidence had indicated that the complex does tend to become unstable and immunohistochemistry staining analysis to detect SDHB becomes negative.
•A double-hit SDH mutation in pheochromocytoma-paraganglioma virtually always has tended to be associated with a germline mutation rather than being caused by two somatic events. In view of this, it has been iterated that negative staining for SDHB does suggest the need for genetic testing for SDHB, SDHC, as well as SDHD in patients who manifest with pheochromocytoma-paraganglioma. [42] [43]
•As of their documentation, one SDHB-negative renal-cell carcinoma in a patient with PGL4 had been reported [42]
•They had undertaken immunohistochemical analysis to detect SDHB in renal-cell carcinoma specimens obtained from three kindreds who were subsequently shown to harbour a germline SDHB mutation [42] [43]. SDHB was completely negative in all three specimens of SDHB mutation-related renal-cell carcinoma, but it was positive in non-neoplastic cells obtained from these patients and in 70 unselected renal neoplasms that were utilized as controls. The unselected renal neoplasms included 45 conventional clear-cell renal carcinomas (including 3 associated with the von Hippel-Lindau syndrome), 10 papillary renal carcinomas, 9 chromophobe carcinomas, 5 oncocytomas, and 1 hereditary leiomyomatosis renal-cell carcinoma-related neoplasm. Given the parallels with pheochromocytoma-paraganglioma, it is possible that negative staining for SDHB may ensue SDHD-mutated renal-cell carcinoma. Negative SDHB staining also occurs in a distinct subgroup of gastrointestinal stromal neoplasms (paediatric wild-type gastrointestinal stromal tumours, the gastrointestinal stromal tumours of the Carney triad, and the sub-group of adult gastrointestinal stromal tumours that these tumours simulate clinically and pathologically). [44] Not all of these gastrointestinal stromal tumours would be associated with identifiable mitochondrial complex II mutations. In view of this, it is likely that there are other as-yet-undescribed mechanisms of mitochondrial complex II instability and tumorigenesis which are also associated with negative staining for SDHB.
•Immunohistochemical studies analysis to identify SDHB could be utilized to screen patients who have kidney tumours for underlying SDHB germline mutations (and possibly other mitochondrial complex II-related syndromes) at a fraction of the time and cost of formal genetic testing.
Ozluk et al. [45] stated the following:
•Renal cell carcinoma (RCC) which is linked to germline mutation of succinate dehydrogenase subunits A, B, C, and D (SDHA, SDHB, SDHC, and SDHD, respectively) had, within the recent past been included as a provisional entity in the 2013 International Society of Urological Pathology Vancouver classification.
•Majority of SDH-deficient tumours had depicted SDHB mutation, with only a small number of RCC with SDHC or SDHD having been reported up to the time of publication of their article.
•Only one case of SDH-deficient renal carcinoma known to be SDHA mutated had been previously documented.
Ozluk et al. [45] reported an additional RCC which harboured an SDHA mutation, which had afflicted a 62-year-old man with the manifestation of right flank pain and nodal metastasis. The tumour was noted to be typified by an infiltrative pattern with solid, acinar, and papillary components. Loss of SDHA and SDHB protein by immunohistochemistry studies of the tumour had established the diagnosis. Hybrid capture-based comprehensive genomic profiling studies undertaken, had identified 3 genomic alterations within the tumour tissue including: (a) a novel single-nucleotide splice site deletion in SDHA gene, (b) single-nucleotide deletion in NF2 gene, and (c) EGFR gene amplification of 19 copies. Ozluk et al. [45] made the ensuing iterations:
•Their reported case was the second report of SDHA-mutated RCC.
•With increased awareness, this rare tumour could be recognized upon the basis of distinctive morphology and confirmation by immunohistochemistry staining studies and genomic profiling.
Aggarwal et al. [46] stated the following:
•Reduced succinate dehydrogenase (SDH) activity resulting in adverse succinate accumulation had been previously considered relevant only in 0.05% to 0.5% of kidney cancers associated with germline SDH mutations.
•They had examined a broader role for SDH loss in kidney cancer pathogenesis/progression.
•They reported that under-expression of SDH subunits resulting in accumulation of oncogenic succinate is a common feature in clear cell renal cell carcinoma (ccRCC), which does account for about 80% of all kidney cancers, with a marked adverse impact upon survival in ccRCC patients in 516) cases they had studied.
•They had demonstrated that SDH down-regulation is a critical brake in the TCA cycle during ccRCC pathogenesis and progression.
•In exploring mechanisms of SDH down-regulation in ccRCC, they had reported that Von Hippel-Lindau loss-induced hypoxia-inducible factor–dependent up-regulation of miR-210 does cause direct inhibition of the SDHD transcript.
•In addition, shallow deletion of SDHB does tend to occur in about 20% of ccRCC.
•They had illustrated that SDH loss-induced succinate accumulation does contribute to adverse loss of 5-hydroxymethylcytosine, gain of 5-methylcytosine, and enhanced invasiveness in ccRCC via inhibition of ten-eleven translocation (TET)-2 activity.
•Intriguingly, binding affinity between the catalytic domain of recombinant TET-2 and succinate was noted to be very low, indicating that the mechanism of succinate-induced attenuation of TET-2 activity is likely by means of product inhibition rather than competitive inhibition.
•Lastly, exogenous ascorbic acid, which is a TET-activating demethylating agent, led to reversal of the above oncogenic effects of succinate in ccRCC cells.
•Collectively, their study had illustrated that functional SDH deficiency is a common adverse feature of ccRCC and it is not just limited to the kidney cancers that are associated with germline SDH mutations.
•It has been documented clear cell renal cell carcinoma (ccRCC) the commonest type of kidney cancer, which accounts for about 80% of all kidney cancers.
•Despite recent advances, metastatic ccRCC has remained a generally incurable malignancy, with a 5-year survival rate of 20%, which highlights the necessitation for additional biological as well as therapeutic insights in this disease.
•The succinate dehydrogenase (SDH) complex is the only enzyme which represents an integral component of both the TCA cycle and the Electron Transport Chain; hence, playing a pivotal role in oxidative phosphorylation.
•SDH does convert succinate to fumarate in the TCA cycle. The complex is located within the inner mitochondrial membrane and is comprised of four subunits: two hydrophilic subunits, SDHA and SDHB, and two hydrophobic membrane which anchor subunits, SDHC and SDHD.
•In their study, utilising bioinformatic analyses of ccRCC TCGA (KIRC) data (transcriptome, methylome, and survival), ccRCC metabolomic repository, primary ccRCC tumours with adjacent normal renal tissue, and an array of mechanistic/representative experiments, they [a] had investigated the expression of SDH subunits in ccRCC and the prognostic and functional consequences of loss of SDH in ccRCC, [b] had shed light upon the mechanisms of down-regulation of SDH in ccRCC, [c] established the role of succinate as an important epigenetic modulating oncometabolite in ccRCC pathogenesis and progression, as well as [d] determined the potential of ascorbic acid (AA) in reversing the oncogenic effects of succinate in ccRCC.
Kumar et al. [47] iterated the ensuing:
•Succinate dehydrogenase (SDH)- deficient renal cell carcinoma (RCC) is a newly identified rare sub-type of RCC, which had only gained acceptance from the World Health Organization in 2016.
•To the best of their knowledge, at the time of the report of their case, there were only 55 reported cases worldwide.
Kumar et al. [47] reported a 49-year-old man who was incidentally found to have a large right kidney mass. He did not have any personal or family history of paragangliomas (PGL), pheochromocytomas (PC), or gastrointestinal stromal tumors (GIST). His neoplasm was unilateral and unifocal. He underwent an open partial nephrectomy. Detailed pathology examination of the neoplasm was undertaken in order to establish the diagnosis. Genetic testing that was undertaken demonstrated a pathogenic mutation in the SDHB gene. He had been followed-up for 24 months by the time of publication of the article, and he had remained well without any evidence of local or distant recurrence. features. Kumar et al. [47] made ensuing summating discussion iterations:
•Without the identification of SDHB deficiency, their patient’s personal and familial predisposition to PC, PGL, GIST and metachronous RCCs might have gone undetected despite his RCC diagnosis.
•When faced with an eosinophilic RCC, pathologists should routinely look for vacuoles or flocculent cytoplasmic inclusions. When these are found to be present in the neoplasm, or in cases of difficult eosinophilic renal tumors, they would recommend that pathologists should undertake staining for SDHB.
•For neoplasms without adverse pathology examination features including: high nuclear grade, coagulative necrosis, or sarcomatoid differentiation, excision of neoplasm alone might be a reasonable option, with the addition of regular surveillance for PC and PGLs in those neoplasms that are found to harbor germline SDH mutations.
Fuchs et al. [48] stated the following:
•Majority of succinate dehydrogenase (SDH)-deficient renal cell carcinomas (RCCs) demonstrate stereotypical morphology which is typified by bland eosinophilic cells with frequent intracytoplasmic inclusions.
•Nevertheless, variant morphological features had been increasingly recognized. They therefore sought to investigate the incidence and characteristics of SDH-deficient RCC with variant morphologies.
Fuchs et al. [48] studied a multi-institutional cohort of 62 new SDH-deficient RCCs from 59 patients. They reported that the median age at manifestation of the patients was 39 years and the age at manifestation of the patients had ranged from 19 years to 80 years, with a slight male predominance with a male to female (M / F:) ratio of 1.6:1. A relevant family history was reported in 9 patients which amounted to 15%. Multifocal or bilateral neoplasms were identified radiologically in 5 patients, which amounted to 8% of the patients. Typical morphology was found to be present at least focally in 59 neoplasms which amounted to 95% of the neoplasms. Variant morphologies were identified in 13 neoplasms which amounted to 21% of the neoplasms and included high-grade nuclear features and various combinations of papillary, solid, and tubular architecture. Necrosis was identified in 13 neoplasms, 7 of which had demonstrated variant morphology. All 62 neoplasms had demonstrated loss of SDHB expression by immunohistochemistry. None of the reported neoplasms had shown loss of SDHA expression. Germline SDH mutations were identified in all 18 patients for whom the results of testing were known. Among patients for whom follow-up data was available, metastatic disease was documented in 9 cases, 8 of whom had necrosis and/or variant morphology in their primary neoplasm. Three patients died of their disease. Fuchs et al. [48] made the ensuing conclusions:
•Variant morphologies and high-grade nuclear features do occur in a sub-set of SDH-deficient RCCs and they are associated with more aggressive biological behaviour.
•They therefore recommend grading all SDH-deficient RCCs and they also emphasized the need for a low threshold for the undertaking of SDHB immunohistochemistry in any difficult to classify renal neoplasm, particularly if the neoplasm occur at a younger age.
Higashi et al. [49] stated that Succinate dehydrogenase (SDH)-deficient renal cell carcinoma (RCC) is a rare cancer of the kidney. Higashi et al. [49] reported a 75-year-old Japanese woman who had manifested with visible haematuria. He had computed tomography scan which demonstrated two neoplasms within tumours in the left kidney, which were resected. Immunohistochemistry staining studies of the neoplasms indicated negative staining for the B subunit of SDH (SDHB) in the resected specimen, leading to a final diagnosis of SDHB-deficient RCC. Genetic testing for SDHB had demonstrated a RCC germline variant in exon 6 (NM 003000.3:c.642 G > C) that which had been previously reported but associated with a novel phenotype (for example., RCC). Twenty-six years earlier, her daughter, who was 25 years old at the time, had undergone radical nephrectomy for a pathology diagnosis of renal oncocytoma of her right kidney; SDHB immunostaining of her daughter’s neoplasm was also negative retrospectively. Higashi et al. [49] had confirmed that her daughter carried the germline variant in SDHB exon 6, which was similar what the patient carried. The patient did not have any evidence of disease progression at 15 months pursuant to her surgery.
Higashi et al. [49] made the ensuing iterations:
•Even though SDH-deficient renal cell carcinoma (RCC) occurs an uncommon tumour, it had been recently identified as a unique sub-type of kidney tumour in the 2016 World Health Organization classification [50]
•Majority of patients who have SDH-deficient RCC do harbour a germline variant in SDH.
•It has been documented that the estimated incidence rate of SDH-deficient RCC among all RCCs is 0.05% to 0.2%. [27].
•Sixty cases of SDH-deficient RCC had been reported in the literature by July 2022 [51]
•Among SDH-deficient RCCs, SDHB-deficient RCC has tended to be the predominant type, whilst SDHA-, SDHC-, and SDHD-deficient RCC are stated to be less common [1] [52]
Higashi et al. [49] reported a 75-year-old Japanese woman who was referred to their hospital with tumours within her left kidney, which were identified during her assessment for visible haematuria. She did not have any significant past medical disease or family history of paraganglioma/pheochromocytoma (PPGL), gastrointestinal stromal tumour, or pituitary adenoma. Twenty-six years earlier, her daughter, who was 25 years old, had undergone right radical nephrectomy, with a pathology examination diagnosis of renal oncocytoma. She had contrast-enhanced computed tomography (CT) scan which demonstrated a 3.8 cm × 2.8 cm tumour within the upper pole and a 1.2 cm × 1.1 cm tumour within the lower pole regions of her left kidney (see figures 1 A, and 1B). There was no evidence of metastasis demonstrated in her staging radiology images. The results of all her assessment haematology and biochemistry blood tests were within normal ranges. She underwent robot-assisted partial nephrectomy, and histopathology examination of the tumour revealed SDH-deficient RCC with Fuhrman grade 2/International Society of Urological Pathology grade 2 (see figure 1 C) Microscopy histopathology examination of the tumour demonstrated that the cells were intermediate to large in size, with cytoplasmic vacuoles containing eosinophilic fluid. The Nuclei were noted to round, and prominent nucleoli and apparent perinuclear halos were absent. Immunostaining staining studies for the B subunit of SDH (SDHB) (Abcam ab4714, clone 21A11AE7, Cambridge, UK) was reported as negative, whereas immunostaining study for scattered inflammatory cells was noted to be positive (see figure 1C). Immunostaining studies for the A subunit of SDH (SDHA) (Abcam ab14715, clone 2E3GC12FB2AE2, Cambridge, UK) was reported as positive (see figure 1C). Furthermore, a genetic test was undertaken for SDHB pursuant to the obtaining of her written consent. DNA was extracted from her renal tumour tissue, normal kidney tissue, and blood utilising a DNeasy blood and tissue kit (Qiagen, Valencia, CA). The primer pairs utilised for exon amplification (exons 1 to 8) of both the tumour as well as normal tissue and DNA sequencing with a 3730XL DNA analyser (Thermo Fisher Scientific, MA USA) were undertaken as had been previously reported [53] Higashi et al. [49] utilised the nucleotide sequence database of SDHB (https://www.ncbi.nlm.nih.gov/nuccore/NG_012340.1) as a normal control. The results demonstrated that the patient carried an RCC germline variant in SDHB exon 6 (NM_003000.3:c.642 G > C) (see figures 2 A, 2 B, and 2 C). During the process of her follow-up clinical and radiology imaging assessments, there was no evidence of disease progression at 15 months pursuant to her surgery at the time of publication of the article. Higashi et al. [49] retrospectively reviewed the patient’s daughter’s renal tumour, which had comprised of eosinophils and oncocytes with multiple cytoplasmic vacuoles; and immunohistochemistry staining studies for SDHB within the tumour lesion was found to be negative (see figure 1 D)..In view of the fact that the patient’s daughter was strongly suspected of being an SDHB variant carrier, her daughter did undergo familial genetic testing of blood following clinical genetic counselling and was she found to carry the same variant in SDHB exon 6 (see figure 2 D).
Figure 1:
Abdominal computed tomography, gross appearance, and microscopic findings: Enhanced computed tomography (CT) showing an enlarged 3.8 x 2.8 cm tumour in the upper pole region of the left kidney and 1.2 x 1.1 cm (arrow) [A] tumour in the lower pole region of the left kidney (arrow) [B] The tumours were well circumscribed with tan-brown [A] and reddish-brown [B] cut surfaces. Histopathological examination revealed SDH-deficient RCC with Fuhrman grade 2 / International society of Urological Pathology grade 2 [C]. Cells were intermediate to large in size with cytoplasmic vacuoles containing eosinophilic fluid. Nuclei were round with prominent nucleoli, and apparent perinuclear halos were absent. Immunostaining for SDHB was negative but positive for scattered inflammatory cells [C]. Immunostaining for SDHA was positive [C]. Histopathology of the daughter’s tumours showed eosinophilic staining for SDHB [D] [bar = 50 µm]. Reproduced from: [49 Higashi S, Sasaki T, Uchida K, Kageyama T, Ikejiri M, Matsumoto R, Kato M, Masui S, Yoshio Y, Nishikawa K, Okugawa Y, Watanabe M, Inoue T. Succinate dehydrogenase B-deficient renal cell carcinoma with a germline variant in a Japanese patient: a case report. Hum Genome Var. 2022 Jul 22;9(1):25. doi: 10.1038/s41439-022-00202-z. PMID: 35869040; PMCID: PMC9307839. Under Copy Right: Copyright © The Author(s) 2022 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third-party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory
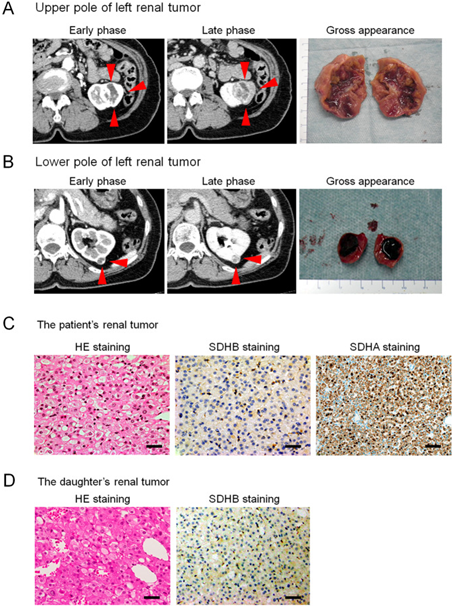
Figure 1: Abdominal computed tomography, gross appearance, and microscopic findings:
Enhanced computed tomography (CT) showing an enlarged 3.8 x 2.8 cm tumour in the upper pole region of the left kidney and 1.2 x 1.1 cm (arrow) [A] tumour in the lower pole region of the left kidney (arrow) [B] The tumours were well circumscribed with tan-brown [A] and reddish-brown [B] cut surfaces. Histopathological examination revealed SDH-deficient RCC with Fuhrman grade 2 / International society of Urological Pathology grade 2 [C]. Cells were intermediate to large in size with cytoplasmic vacuoles containing eosinophilic fluid. Nuclei were round with prominent nucleoli, and apparent perinuclear halos were absent. Immunostaining for SDHB was negative but positive for scattered inflammatory cells [C]. Immunostaining for SDHA was positive [C]. Histopathology of the daughter’s tumours showed eosinophilic staining for SDHB [D] [bar = 50 µm]. Reproduced from: [49 Higashi S, Sasaki T, Uchida K, Kageyama T, Ikejiri M, Matsumoto R, Kato M, Masui S, Yoshio Y, Nishikawa K, Okugawa Y, Watanabe M, Inoue T. Succinate dehydrogenase B-deficient renal cell carcinoma with a germline variant in a Japanese patient: a case report. Hum Genome Var. 2022 Jul 22;9(1):25. doi: 10.1038/s41439-022-00202-z. PMID: 35869040; PMCID: PMC9307839. Under Copy Right: Copyright © The Author(s) 2022 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third-party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
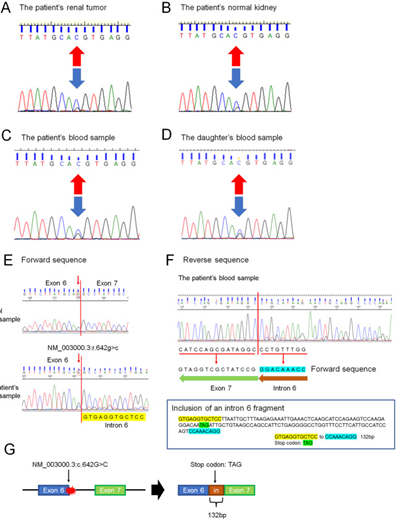
Figure 2: PCR directed sequencing of SDHB exon 6.
DNA from the patient’s tumour tissue showed a variant in RCC (NM 00300.3: c.642>C) arrow previously reported but associated with a novel phenotype (RCC) (A). DNA from the patient’s normal tissue and blood sample showed some variant (NM 003000.3: c.642 G>C) (arrow) (B and C). DNA from the daughter’s blood sample showed the same variant (NM 003000.3:c642 G > C) (arrow) (D). Effect of the SDHB variant (NM 003000.3: c.642 G > C) on splicing by RNA analysis. Forward sequence data of cloned RT-PCR products revealed that the 3’ splice site of SDHB exon 6 was not recognized, with aberrant transcription continuing into intron 6 (E). Reverse sequence data of cloned PT-PCR products revealed the presence of 132 base pairs of intronic sequences adjacent to exon 7, indicating intron 6 retention (F). The intronic sequence included a stop codon (TAG) (F). The SDHB variant (NM 003000.3:c.642 G > C) results in usage of the intronic spice site, leading to the inclusion of an intron fragment (132 base pairs) (NM 003000.3:r[642 g > c;642_643ins642 + 1_642 + 132]) including a stop codon (TAG) (G), which may be a protein-truncating variant (NM 003000.3:p. Gln214delinsHisValArgCysSerLeuIleAlaLeuArgGluIleGluThrGlnAlaSerArgSerProArgGlyGlnTer) Reproduced from: [49] Higashi S, Sasaki T, Uchida K, Kageyama T, Ikejiri M, Matsumoto R, Kato M, Masui S, Yoshio Y, Nishikawa K, Okugawa Y, Watanabe M, Inoue T. Succinate dehydrogenase B-deficient renal cell carcinoma with a germline variant in a Japanese patient: a case report. Hum Genome Var. 2022 Jul 22;9(1):25. doi: 10.1038/s41439-022-00202-z. PMID: 35869040; PMCID: PMC9307839.Under Copy Right: Copyright © The Author(s) 2022 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third-party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Higashi et al. [49] made the ensuing educative discussions:
•Previous studies had summated the distribution of SDHB germline variants in RCC; [1] [54], nevertheless, the variant that had been identified in their study had not been previously reported in RCC. Notably, the same SDHB variant (NM_003000.3: c.642 G > C) had been reported in patients who had malignant paraganglioma8. 8. [55]
•Based upon the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar), the pathogenic meaning of this SDHB variant is not certain. However, based upon the OncoKB database (https://www.oncokb.org), the amino acid change was located within the 4Fe-4S cluster domain, which binds iron-sulfur clusters.
•Saxena et al. [56] had summarized missense SDHB variants that cause familial cancer syndromes and indicated that the 4Fe–4S cluster domain is the most common site of missense variants [56]
•Furthermore, as the patient’s variant had involved the last nucleotide of exon 6 of the SDHB coding sequence, they had investigated the effect of this SDHB variant on splicing by RNA analysis. They undertook RNA extraction of blood samples and had carried out generated cDNA. cDNA PCR amplification utilising the primers forward (Fw) primer 5′-CAGACAAGGCTGGAGACAAACC-3′ and reverse (Rv) primer 5′-GCAATAGCTTTCCCTGGATTCAGAC-3′ for targeting SDHB exon 3 to exon 8. Higashi et al. [49] analysed the cloned RT–PCR products by means of direct sequencing, which revealed that the 3′ splice site of SDHB exon 6 was not recognized, with aberrant transcription continuing into intron 6 (see figure 2 E). Reverse sequence data of the cloned RT–PCR products had demonstrated the presence of 132 base pairs of intronic sequence adjacent to exon 7, which indicated retention of intron 6 (see figure 2F). The intronic sequence included a stop codon (TAG) (see figure 2 F). Hence, the SDHB variant (NM_003000.3:c.642 G > C) results in usage of the intronic splice site, leading to the inclusion of an intron fragment (132 base pairs) (NM_003000.3: r.[642 G > C;642_643ins642 + 1_642 + 132]) (see figure 2 G).which might constitute a protein-truncating variant (NM_003000.3:p.Gln214delinsHisValArgCysSerLeuIleAlaLeuArgGluIleGluThrGlnAlaSerArgSerProArgGlyGlnTer).
•Based upon their findings, the patient’s and her daughter’s variant (NM_003000.3:c.642 G > C) could be a pathogenic change.
•SDH is well-recognized as a tumour suppressor.
•When double-hit inactivation does occur within SDHx (typically with one germline and somatic variant), the entire complex does become unstable, emanating in degradation of the B subunit.
•Andrews et al. [57] had found that the risk for the development of developing a renal tumour by the age of 60 years in SDHB variant carriers was 4.2% (95% CI 0.46–7.8%) by Kaplan–Meier analysis of nonprobands and 4.71% (95% CI 1.65–7.7%) by Kaplan–Meier analysis of all SDHB variant carriers. [57]
•SDH-deficient RCC generally does occurs in young adulthood (i.e., median 36.8 years [range, 14–76 years]), [58] even though the optimal initial evaluation and follow-up of asymptomatic carriers of SDHx variants had not yet been agreed upon. However, germline mutations within SDHx genes are responsible for about 20% of cases of PPGL and had been stated to also be associated with the presence of other SDHx-related tumours. [59] Hence, PPGL evaluation might have positive effects upon the outcomes, including survival.
•The risk for the development of RCC had been iterated to be smaller in comparison with that of PPGL for SDHx gene variant carriers, and therefore, abdominal radiology imaging incorporating renal imaging for RCC evaluation might suffice for carriers.[59]
•The morphology of SDHB-deficient RCC does overlap with various patterns of histology subtypes, including: chromophobe RCC, clear cell RCC, papillary RCC, sarcomatoid RCC, unclassified RCC, as well as oncocytoma of kidney. [60]
•It has been pointed out that both oncocytoma and SDHB-deficient RCC demonstrate tumour cells that contain uniformly round nuclei and eosinophilic cytoplasm and are arranged in solid nests, acini, tubules, or a cystic pattern. [58]
•However, intra-tumoral mast cells and cytoplastic vacuoles tend to be rarely visualised in oncocytoma, and the cytoplasm of SDHB-deficient RCC tends to be flocculent but not truly oncocytic in nature. [58]
•In the case of the existence of a family history, renal oncocytoma in young adulthood needs to be differentiated from SDH-deficient RCC.
•Even though SDHB-deficient RCC does demonstrate a strong correlation with germline SDH variants, immunohistochemistry staining study is a powerful tool to ascertain a patient’s phenotype, as opposed to genetic testing.
•Immunohistochemically, tumour cells in SDHB-, SDHC-, and SDHD-deficient RCC tend to be negative for SDHB but positive for SDHA.
•It has been iterated that loss of SDHB staining does indicate disruption of SDH complex II2. On the contrary, it has been documented that tumour cells tend to be negative for both SDHA and SDHB in SDHA-deficient RCC1. [50]
•The reason why the SDHA protein does remain stable in the presence of SDHB, SDHC, or SDHD variants is not known or explained. [50]
•In their reported case, they did not have any information regarding an additional SDHB hit for SDHB deficiency.
•According to Knudson’s two-hit model postulation, loss of heterozygosity (LOH) or copy number change, deep intronic change disrupting another SDHB allele, or epigenetic dysregulation within the tumour might have occurred during tumorigenesis in their reported patient. Furthermore, the mechanisms of differential onset of SDH-deficient RCC in their reported patient and her daughter had remained uncleared.
Williamson et al. [31] iterated the following:
•Patients who have germline mutation of succinate dehydrogenase (SDH) subunit genes are prone to the development of paraganglioma, gastrointestinal stromal tumour, and on rare occasions renal cell carcinoma (RCC).
•Nevertheless, SDH-deficient RCC had not yet been widely recognized.
Williamson et al. [31] identified such tumours by distinctive morphology and confirmed absence of immunohistochemistry staining for SDHB. Immunohistochemistry staining features were evaluated by Williamson et al. [31] utilising a panel of antibodies to renal tumour antigens. Targeted next-generation sequencing was undertaken upon DNA extracted from paraffin-embedded tissue. Williamson et al. [31] identified eleven tumours from 10 patients, whose median age was 40 years and whose ages had ranged between 22 years and 72 years. Williamson et al. [31] reported the following:
•Two patients had paragangliomas, 1 patient had bilateral SDH-deficient RCC, and 1 patient had contralateral oncocytoma.
•Grossly, the tumours were found to be tan or red-brown, 2 cm to 20 cm in diameter and their median diameter was 4.25 cm.
•Fuhrman grades of the tumours were reported to be 2 in 10 cases or 3 in 1 case.
•Stage the tumours was pT1a-pT2b.
•One patient had developed widespread metastases 16 years after undergoing of nephrectomy and died of disease 6 years later.
•All tumours were documented to be composed of uniform eosinophilic cells containing vacuoles or flocculent cytoplasmic inclusions.
•The architecture of the tumour was reported to be primarily solid; entrapped renal tubules and intratumoral mast cells were noted to be common.
•Based upon immunohistochemistry studies, the tumour cells were found to have exhibited negative staining for SDHB in 11 out of 11 tumours and rarely SDHA in 1 out of 11 tumours. Labelling was reported to be uniformly positive for PAX8 and kidney-specific cadherin and labelling was absent for KIT, RCC, and carbonic anhydrase IX. Staining for broad-spectrum epithelial markers was found to be often negative or focal (positive staining for AE1/AE3 in 4/10, CAM5.2 3/7, CK7 1/11, EMA 10/10). By sequencing, SDHB mutation and loss of the second allele were noted to be present in 5 out of 6 tumours; the SDHA-deficient tumour had demonstrated no SDHB abnormality.
•SDH-deficient RCC is a unique neoplasm which is capable of progression, often harbouring SDHB mutation.
•A monomorphic oncocytic kidney tumour with solid architecture, cytoplasmic inclusions of flocculent material, and intratumoral mast cells should prompt evaluation of SDH status, as it might have implications for screening the patient and relatives.
•Negative immunohistochemistry for KIT and heterogeneous labelling for epithelial antigens are other supportive features.
Gill et al. [27] iterated that Succinate dehydrogenase (SDH)-deficient renal carcinoma had been accepted as a provisional entity in the 2013 International Society of Urological Pathology Vancouver Classification. In order to further define the morphological and clinical features of SDH, Gill et al. [27] studied a multi-institutional cohort of 36 SDH-deficient renal carcinomas from 27 patients, which included 21 previously unreported cases. Gill et al. [27] estimated that 0.05% to 0.2% of all renal carcinomas are SDH deficient. Gill et al. [27] also stated that the mean age of the patients at the time of their presentation was 37 years and the ages at their initial presentation had ranged between 14 years 76 years, with a slight male predominance of 1.7 to 1 (Male to Female = 1.7:1). Gill et al. [27] observed bilateral tumours in 26% of the patients. Thirty-four tumours that amounted to 94% of the tumours had demonstrated the previously reported morphology at least focally, which included: solid or focally cystic growth, uniform cytology with eosinophilic flocculent cytoplasm, intracytoplasmic vacuolations and inclusions, and round to oval low-grade nuclei. All 17 patients who underwent genetic testing for mutation in the SDH subunits had demonstrated germline mutations with 16 in SDHB and 1 in SDHC. Nine out of 27 patients which amounted to 33% of the patients had developed metastatic disease, 2 of them after prolonged follow-up (5.5 years and 30 years). Seven of 10 patients which amounted 70% of the patients with high-grade nuclei metastasized as did all 4 patients who had coagulative necrosis. Two of 17 that amounted to 12% of patients with low-grade nuclei metastasized, and both had unbiopsied contralateral tumours, which might have been the origin of the metastatic disease.
Gill et al. [27] made the ensuing conclusions:
•SDH-deficient renal carcinoma is an uncommon and unique type of renal carcinoma, exhibiting stereotypical morphology features in the great majority of cases and showing a strong relationship with SDH germline mutation.
•Even though this SDH tumour may undergo dedifferentiation and metastasize, sometimes after a prolonged delay, metastatic disease is rare in the absence of high-grade nuclear atypia or coagulative necrosis.
Conclusions:
•Succinate dehydrogenase-deficient RCC is an uncommon subtype of RCC, which does generally tend to portend an indolent biological behaviour.
•Even though uncommon, it is pivotal to identify SDH-deficient RCC in view of the strong association with syndromic disease including most commonly paraganglioma/pheochromocytomas.
•Succinate dehydrogenase-deficient RCC has important implications for the patient and the family, in view of the fact that they require counselling and assessment and genetic testing with regular follow-up for syndromic disease.
•The poor prognostic factors of Succinate dehydrogenase-deficient RCC should be clearly understood by all clinicians, pathologists and Urologists so that high-risked tumours are treated aggressively including regular long-term clinical and radiology imaging follow-up.
•A global multi-centre trial of high-risk Succinate dehydrogenase-deficient RCCs should be commenced in order to ascertain the best treatment options for these tumours including immunotherapy, radiotherapy, combination chemotherapy as well as other available treatment options that can be suggested.
Conflict Of Interest
None
Acknowledgements
Acknowledgements to:
•Human Genome Variation for granting permission for reproduction of figures and contents of their excellent educative article under Copy Right: Copyright © The Author(s) 2022 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third-party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
References
- Ricketts CJ, Shuch B, Vocke CD, Metwalli AR, Bratslavsky G, et al. (2012). Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. J Urol;188(6):2063-2071.
View at Publisher | View at Google Scholar - Linehan WM, Srinivasan R, Schmidt LS. (2010). The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol;7(5):277-285.
View at Publisher | View at Google Scholar - Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, et al. (2002). Multiple Leiomyoma Consortium. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. Apr;30(4):406-410. 1186-5300.
View at Publisher | View at Google Scholar - Grubb RL 3rd, Franks ME, Toro J, Middelton L, Choyke L, et al. (2007). Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol;177(6):2074-2079; discussion 2079-2080.
View at Publisher | View at Google Scholar - Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, et al. (2003). Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet.;73(1):95-106.
View at Publisher | View at Google Scholar - Warburg O, Posener K, Negelein E. Metabolism of the Carcinoma Cell. Biochem Z. 1924; 152:309
View at Publisher | View at Google Scholar - Buchanan JW. (1927).SOME LIMITATIONS OF WARBURG'S THEORY OF THE ROLE OF IRON IN RESPIRATION. Science;66(1706):238-239.
View at Publisher | View at Google Scholar - Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, et al. (2010). UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet;196(1):45-55.
View at Publisher | View at Google Scholar - Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, et al. (2011). The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell.;20(3):315-327.
View at Publisher | View at Google Scholar - Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peçzkowska M, et al. (2004). Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet;74(1):153-159.
View at Publisher | View at Google Scholar - Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, et al. (2000). Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science.;287(5454):848-851.
View at Publisher | View at Google Scholar - Niemann S, Müller U. (2000). Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet;26(3):268-270.
View at Publisher | View at Google Scholar - Astuti D, Douglas F, Lennard TW, Aligianis IA, Woodward ER, et al. (2001). Germline SDHD mutation in familial phaeochromocytoma. Lancet.;357(9263):1181-1182.
View at Publisher | View at Google Scholar - Burnichon N, Brière JJ, Libé R, Vescovo L, Rivière J, et al. (2010). SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet.;19(15):3011-3020
View at Publisher | View at Google Scholar - Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, et al. (2008). Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet.;16(1):79-88.
View at Publisher | View at Google Scholar - Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P, et al. (2011). NIH Pediatric and Wild-Type GIST Clinic; O'Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L, Stratakis CA. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A;108(1):314-318.
View at Publisher | View at Google Scholar - Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, et al (2004). European-American Paraganglioma Study Group. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA;292(8):943-951.
View at Publisher | View at Google Scholar - Srirangalingam U, Walker L, Khoo B, MacDonald F, Gardner D, et al. (2008). Clinical manifestations of familial paraganglioma and phaeochromocytomas in succinate dehydrogenase B (SDH-B) gene mutation carriers. Clin Endocrinol (Oxf);69(4):587-596.
View at Publisher | View at Google Scholar - Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, et al. (2008). Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst;100(17):1260-1262.
View at Publisher | View at Google Scholar - Schimke RN, Collins DL, Stolle CA. (2010). Paraganglioma, neuroblastoma, and a SDHB mutation: Resolution of a 30-year-old mystery. Am J Med Genet A;152A (6):1531-1535
View at Publisher | View at Google Scholar - Housley SL, Lindsay RS, Young B, McConachie M, Mechan D, et al. (2010). Renal carcinoma with giant mitochondria associated with germ-line mutation and somatic loss of the succinate dehydrogenase B gene. Histopathology;56(3):405-408.
View at Publisher | View at Google Scholar - Henderson A, Douglas F, Perros P, Morgan C, Maher ER. (2009). SDHB-associated renal oncocytoma suggests a broadening of the renal phenotype in hereditary paragangliomas. Fam Cancer;8(3):257-260.
View at Publisher | View at Google Scholar - Gill AJ, Pachter NS, Clarkson A, Tucker KM, Winship IM, et al. (2011). Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome type 4. N Engl J Med. 3;364(9):885-886.
View at Publisher | View at Google Scholar - Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, et al. (2008). Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet;83(2):261-268.
View at Publisher | View at Google Scholar - Malinoc A, Sullivan M, Wiech T, Schmid KW, Jilg C, et al. (2012). Biallelic inactivation of the SDHC gene in renal carcinoma associated with paraganglioma syndrome type 3. Endocr Relat Cancer. May 3;19(3):283-290
View at Publisher | View at Google Scholar - Andeen NK, Tretiakova M. (2016).Kidney tumor Adult renal cell carcinoma. – rare succinate dehydrogenase deficient. Last Author Update.; Last Staff Update 17 March 2023
View at Publisher | View at Google Scholar - Gill AJ, Hes O, Papathomas T, Šedivcová M, Tan PH, et al. (2014). Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol;38(12):1588-1602.
View at Publisher | View at Google Scholar - Cardaci S, Ciriolo M R. (2012). Redox Status and Bioenergetics Special Issue. Liaison in Cancer and Neurodegeneration. Review Article. TCA Cycle Defects and Cancer: When Metabolism Tunes Redox State. International Journal of Cell Biology.
View at Publisher | View at Google Scholar - Else T, Greenberg S, Fishbein L. (2023). Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023.
View at Publisher | View at Google Scholar - Paik JY, Toon CW, Benn DE, High H, Hasovitz C, et al. (2014). Renal carcinoma associated with succinate dehydrogenase B mutation: a new and unique subtype of renal carcinoma. J Clin Oncol. 20;32(6): e10-13.
View at Publisher | View at Google Scholar - Williamson SR, Eble JN, Amin MB, Gupta NS, Smith SC, et al. (2015). Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol;28(1):80-94.
View at Publisher | View at Google Scholar - Zhao W, Tian B, Wu C, Peng Y, Wang H, et al. (2015). DOG1, cyclin D1, CK7, CD117 and vimentin are useful immunohistochemical markers in distinguishing chromophobe renal cell carcinoma from clear cell renal cell carcinoma and renal oncocytoma. Pathol Res Pract;211(4):303-307.
View at Publisher | View at Google Scholar - Michalova K, Tretiakova M, Pivovarcikova K, Alaghehbandan R, Perez Montiel D, et al. (2020). Expanding the morphologic spectrum of chromophobe renal cell carcinoma: A study of 8 cases with papillary architecture. Ann Diagn Pathol; 44:151448.
View at Publisher | View at Google Scholar - Epstein JI, Egevad L, Humphrey PA, Montironi R; (2014). Members of the ISUP Immunohistochemistry in Diagnostic Urologic Pathology Group. Best practices recommendations in the application of immunohistochemistry in the prostate: report from the International Society of Urologic Pathology consensus conference. Am J Surg Pathol;38(8): e6-e19. 25029122.
View at Publisher | View at Google Scholar - Iczkowski KA, Czaja RC. (2019). Eosinophilic Kidney Tumors: Old and New. Arch Pathol Lab Med. 2019;143(12):1455-1463.
View at Publisher | View at Google Scholar - Ng KL, Ellis RJ, Samaratunga H, Morais C, Gobe GC, et al. (2019). Utility of cytokeratin 7, S100A1 and caveolin-1 as immunohistochemical biomarkers to differentiate chromophobe renal cell carcinoma from renal oncocytoma. Transl Androl Urol;8(Suppl 2): S123-S137.
View at Publisher | View at Google Scholar - Liu YJ, Ussakli C, Antic T, Liu Y, Wu Y, et al. (2020). Sporadic oncocytic tumors with features intermediate between oncocytoma and chromophobe renal cell carcinoma: comprehensive clinicopathological and genomic profiling. Hum Pathol.; 104:18-29. 3267-3684.
View at Publisher | View at Google Scholar - Miettinen M, Sarlomo-Rikala M, McCue P, Czapiewski P, Langfort R, et al. (2014). Mapping of succinate dehydrogenase losses in 2258 epithelial neoplasms. Appl Immunohistochem Mol Morphol;22(1):3136
View at Publisher | View at Google Scholar - Gill AJ, Pachter NS, Chou A, Young B, Clarkson A, et al. (2011). Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol;35(10):1578-1585.
View at Publisher | View at Google Scholar - Nathanson K, Baysal BE, Drovdlic C, Komminoth P, Neumann HP. (2004).Familial paraganglioma-phaeochromocytoma syndromes caused by SDHB, SDHC and SDHD mutations. Pathology and Genetics of Tumours of Endocrine Organs (IARC WHO Classification of Tumours Volume 8). World Health Organization. Lyon, France. 238 – 242
View at Publisher | View at Google Scholar - Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010 Jan;31(1):41-51.
View at Publisher | View at Google Scholar - van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, et al. (2009). An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol.;10(8):764-71.
View at Publisher | View at Google Scholar - Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, et al. (2010). Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol;41(6):805-814.
View at Publisher | View at Google Scholar - Gill AJ, Chou A, Vilain R, Clarkson A, Lui M, et al. (2010). Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol;34(5):636-644.
View at Publisher | View at Google Scholar - Ozluk Y, Taheri D, Matoso A, Sanli O, Berker NK, et al. (2015) Renal carcinoma associated with a novel succinate dehydrogenase A mutation: a case report and review of literature of a rare subtype of renal carcinoma. Hum Pathol;46(12):1951-1955.
View at Publisher | View at Google Scholar - Aggarwal RK, Luchtel RA, Machha V, Tischer A, Zou Y, et al. (2021). Functional succinate dehydrogenase deficiency is a common adverse feature of clear cell renal cancer. Proc Natl Acad Sci U S A. 28;118(39)
View at Publisher | View at Google Scholar - Kumar R, Bonert M, Naqvi A, Zbuk K, Kapoor A. (2018). SDH-deficient renal cell carcinoma - clinical, pathologic and genetic correlates: a case report. BMC Urol. 27;18(1):109.
View at Publisher | View at Google Scholar - Fuchs, T.L., Maclean, F., Turchini, J. et al. (2022). Expanding the clinicopathological spectrum of succinate dehydrogenase-deficient renal cell carcinoma with a focus on variant morphologies: a study of 62 new tumors in 59 patients. Mod Pathol; 35, 836–849.
View at Publisher | View at Google Scholar - Higashi S, Sasaki T, Uchida K, Kageyama T, Ikejiri M, et al. (2022). Succinate dehydrogenase B-deficient renal cell carcinoma with a germline variant in a Japanese patient: a case report. Hum Genome Var. 22;9(1):25.
View at Publisher | View at Google Scholar - Wang G, Rao P. (2018). Succinate Dehydrogenase-Deficient Renal Cell Carcinoma: A Short Review. Arch Pathol Lab Med;142(10):1284-1288.
View at Publisher | View at Google Scholar - Szymanski M, Rusetska N, Jancewicz I, Armatowska A, Ligaj M, et al. (2021). Succinate Dehydrogenase-Deficient Renal Cancer Featuring Fructose-1,6-Biphosphatase Loss, Pyruvate Kinase M2 Overexpression, and SWI/SNF Chromatin Remodeling Complex Aberrations: A Rare Case Report. Oncologist.;26(9): e1652-e1655.
View at Publisher | View at Google Scholar - Kamai T, Higashi S, Murakami S, Arai K, Namatame T, et al. (2021). Single nucleotide variants of succinate dehydrogenase A gene in renal cell carcinoma. Cancer Sci.;112(8):3375-3387.
View at Publisher | View at Google Scholar - Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, et al. (2001). Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet;69(1):49-54.
View at Publisher | View at Google Scholar - Iwashita H, Okudela K, Matsumura M, Yamanaka S, Sawazumi T, et al. (2017). Succinate dehydrogenase B-deficient renal cell carcinoma: A case report with novel germline mutation. Pathol Int;67(11):585-589.
View at Publisher | View at Google Scholar - Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, et al. (2006). High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab.;91(11):4505-4509.
View at Publisher | View at Google Scholar - Saxena N, Maio N, Crooks DR, Ricketts CJ, Yang Y, et al. (2016). SDHB-Deficient Cancers: The Role of Mutations That Impair Iron Sulfur Cluster Delivery. J Natl Cancer Inst. Jan;108(1): djv287.
View at Publisher | View at Google Scholar - Andrews KA, Ascher DB, Pires DEV, Barnes DR, Vialard L, et al. (2018). Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet. Jun;55(6):384-394
View at Publisher | View at Google Scholar - Tsai TH, Lee WY. (2019). Succinate Dehydrogenase-Deficient Renal Cell Carcinoma. Arch Pathol Lab Med;143(5):643-647.
View at Publisher | View at Google Scholar - Amar L, Pacak K, Steichen O, Akker SA, Aylwin SJB, et al. (2021). International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endocrinol;17(7):435-444.
View at Publisher | View at Google Scholar - Kuroda N, Yorita K, Nagasaki M, Harada Y, Ohe C, et al. (2016). Review of succinate dehydrogenase-deficient renal cell carcinoma with focus on clinical and pathobiological aspects. Pol J Pathol. 67(1):3-7.
View at Publisher | View at Google Scholar
