

Research | DOI: https://doi.org/10.31579/2835-8465/019
Ekman-Lonstein syndrome in Iraq: An educational article and expert opinion
Advisor in Pediatrics and Pediatric Psychiatry Children Teaching Hospitalof Baghdad MedicalCity iraq.
*Corresponding Author: Aamir Jalal Al Mosawi, Advisor in Pediatrics and Pediatric Psychiatry Children Teaching Hospital of Baghdad Medical City Iraq.
Citation: Aamir Jalal Al Mosawi, Ekman-Lobstein syndrome in Iraq: An educational article and expert opinion, (2024), Orthopedic case reports; 3(6): DOI: 10.31579/2835-8465/019
Copyright: ©, 2024, Aamir Jalal Al Mosawi, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received: 09 December 2024 | Accepted: 18 December 2024 | Published: 25 December 2024
Keywords: ekman-lobstein syndrome; clinical and radiologic features; therapeutic options
Abstract
Background: Ekman-Lonstein syndrome is a genetic bone disorder that has also been called Lobstein syndrome, Vrolik syndrome, Fragilitas ossium,Glass-bone disease, and Brittle bone disease and 0steogenesis imperfecta. The condition is not a single disease, but a variety of disorders or types of variable severity. We have previously described a case of Ekman-Lobstein syndrome in a boy with blue sclerae that was most likely had type 1 of the syndrome according to David Owen Sillence and colleagues’ classification.
Materials and methods: The case of two sisters with chronic deforming bone disease from Maysan Governorate in the south of Iraq, whom were observed during May, 2023 is described.
Results: The older thirteen years old sisterhas significant bowing of the limbs that markedly reduced her mobility, and she was using a wheelchair. The younger sister was at about the age of four and half years, and she had less severe bowing and she was still able to walk. Both girls had history of fracture of the upper limb. Both girls received the diagnosis of refractory rickets and were treated with vitamin D and calcium supplements. Both sisters have no blush sclera. The younger sister laboratory tests performed during May, 2023 showed normal serum calcium of 9 mg/dL and normal serum phosphorus of 5.16 mg/dL. However, the alkaline phosphatase was high at 164 u/l (Normally less than 140 u/l). Radiographs of both sisters showed no rachitic changes, but showed changes consistent with diagnosis of Ekman-Lobstein syndrome.
Conclusion: This paper documented the occurrence of another type of Ekman-Lobstein syndrome in Iraq without blue sclerae, in addition to type 1 (David Owen Sillence and colleagues’ classification) which we have previously reported. The current expert opinion suggests that bisphosphonates have been shown to be beneficial in improving the condition and therefore should be considered as the treatment of choice.
Introduction
Ekman-Lobstein syndrome is a genetic bone disorder that has also been called Lobstein syndrome, Vrolik syndrome, Fragilitas ossium, Glass-bone disease, and Brittle bone disease and 0steogenesis imperfecta.The condition is not a single disease, but a variety of disorders or types of variable severity associated with defective connective tissue formation and deficiency of type-I collagen resulting from an amino acid substitution of glycine to bulkier amino acids in the collagen triple helix structure.The cardinal feature of Ekman-Lobstein syndrome is osteopenia because of bone resorption whichis generally associated with increased alkaline phosphatase resulting clinically in fragile, easily broken bones. Bluish discoloration of the sclerae of the eyes is an important feature of the condition, but it is not present in all types.
In 1788, the Swedish doctor Olof Jakob Ekman described the condition in his doctoralthesis and mentioned cases with the condition observedas early as 1678. In 1833, Jean Lobstein (Figure-1A) studied the condition and differentiated it from osteomalacia. He called the disease "idiopathic osteopsathyrosis.

Figure-1A: Jean GeorgesChrétien Frédéric Martin Lobstein (8 May 8, 1777-March 7, 1835), a German-born, French pathologis
In 1849, Willem Vrolik(Figure-1B) described the condition in a newborninfant with poorly ossified calvarium, and called it osteogenesis imperfecta.

Figure-1B: Willem Vrolik(April 29, 1801-December 22, 1863), a Dutch anatomist and pathologist from Amsterdam
The earliest classification of Ekman-Lobstein syndrome included two main types [1,2]:
Osteogenesis imperfecta tarda which is the less severe type,and fractures are not present at birth.
Osteogenesis imperfecta congenita, a more severe form, and is associated with fractures at birth.
However, severalsubtypes have been proposedduring the previousdecades [1,2].
In 1979, David Owen Sillence (Figure-1C) from Australia and his colleagues emphasized the genetic heterogeneity of Ekman-Lobstein syndrome, and reported a genetic epidemiological study which showedthat there are at least four distinct types of syndromes. The autosomal dominantsyndrome (Type- 1) was found to be the most common and is associated with fractures and definite blue sclerae, and can also be associated with pre-senile conductive deafness.

Figure-1C:David Owen Sillence, a geneticist from Australia
A severe type associated with early neonatal fractures and early death was transmitted as an autosomal recessive condition in some, but not all the cases (Type-2).
A moderately severe, mostly sporadic type with about two thirds of the affected patients experience fractures at birth and severe limbs and spine progressive deformity. Scleral blueness is less definitethan in the commoner autosomal dominant type and may decrease with age. This type may also appear as a dominant and recessive condition (Type-3).
The fourth type with normal sclerae has autosomal dominant transmission and is associated with fractures, with variable long bones deformity [4].
We have previously described a case of Ekman-Lobstein syndrome in a boy with blue sclerae that was most likelyhad type 1 of the syndrome according to David Owen Sillence and colleagues’ classification [1,2].
Materials and Methods
The case of two sisters with chronic deforming bone disease from Maysan Governorate in the south of Iraq, whom were observed during May, 2023 is described.
The older thirteen years old sister has significant bowing of the limbs that markedly reducedher mobility, and she was using a wheelchair. The younger sister was at about the age of four and half years, and she had less severe bowing and she was still able to walk.
Both girls had history of fracture of the upper limb. Both girls received the diagnosis of refractory rickets and were treated with vitamin Dand calcium supplements.
Results
The family explained that they lost the hope in improving the older sister, and they emphasized that they came to Baghdad with hope that some intervention can prevent the younger sisterfrom becoming like her older sister. They stopped treating the older sister long ago after her illness was worsening despiteconsulting several doctors in Maysan Governorate and in Basra Governorate, and despite treating her for few years. They started treating the younger sister at about the age of 18 months and have stopped treating her before few months after experiencing a fracture after a minor injury. The parents were close relatives, and there was no other affected member in the family. Both sistershave no blush sclera.
The younger sister laboratory tests performed during May, 2023 showed normal serum calcium of 9 mg/dL and normal serum phosphorus of 5.16 mg/dL. However, the alkaline phosphatase was high at 164 u/l (Normally less than 140 u/l).
Old radiographs of the older sister showed osteopenia, fracture in the upper limb, and progressive bowing with no rachitic changes. Figure-2A and B shows a lower limb radiograph taken during December, 2019. Later radiographs showed more severe bowing (Figure-2C).
Old radiographs of the younger sister showed osteopenia, fracture in the upper limb, and progressive bowingwith no rachitic changes. Figure-3A and 3B show lower limb radiographs showing progressive bowing.
Figure-4 (A, B, C, and D) show recentbone radiographs of the youngersister taken during May, 2023 which showed osteopenia, bowing with no rachitic changes. Radiograph of the pelvis showed mild protrusio acetabula (Figure- 4A). Radiograph of the wrist showed no rachitic changes (Figure-4B). Radiograph of the lower limbs showed bowing,cortical thinning with scanty spongiosa (Figure-4C). Lateralskull radiographs showedplatybasia (Figure- 4D).
Therefore, the diagnosis of Ekman-Lobstein syndrome without blue sclerae was made, and it was most likely type 4 according David Owen Sillence [3] and colleagues’ classification despite the inheritance seems more likely to autosomal recessive.
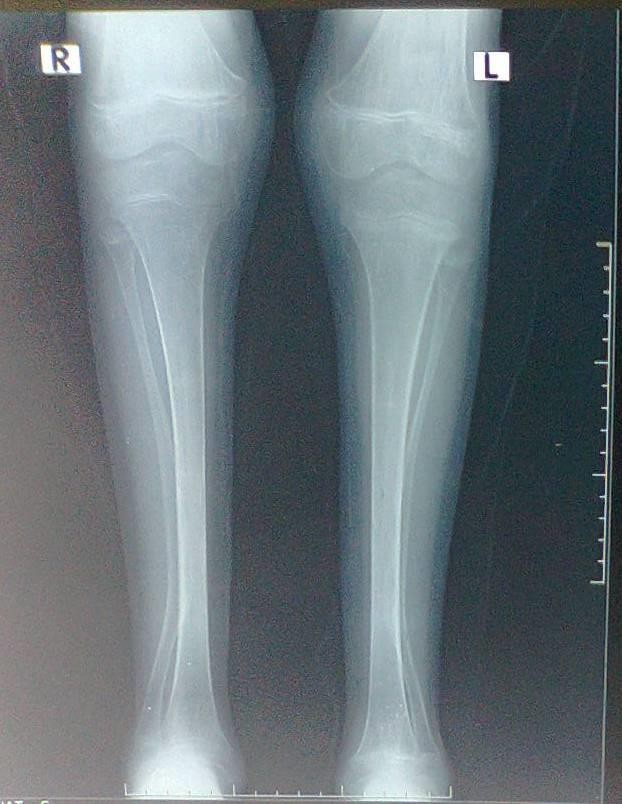
Figure-2A:A lower limb radiograph taken during December,2019
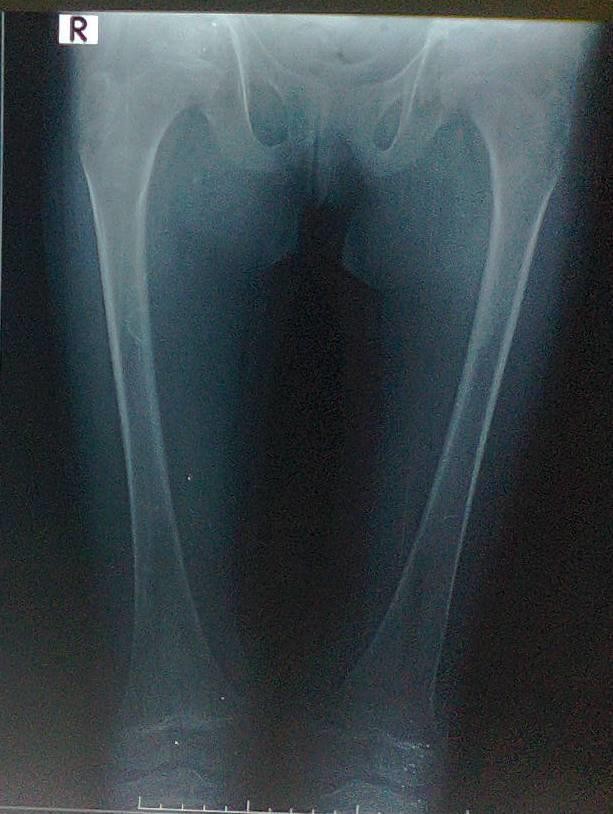
Figure-2B:A lower limb radiograph taken during December, 2019
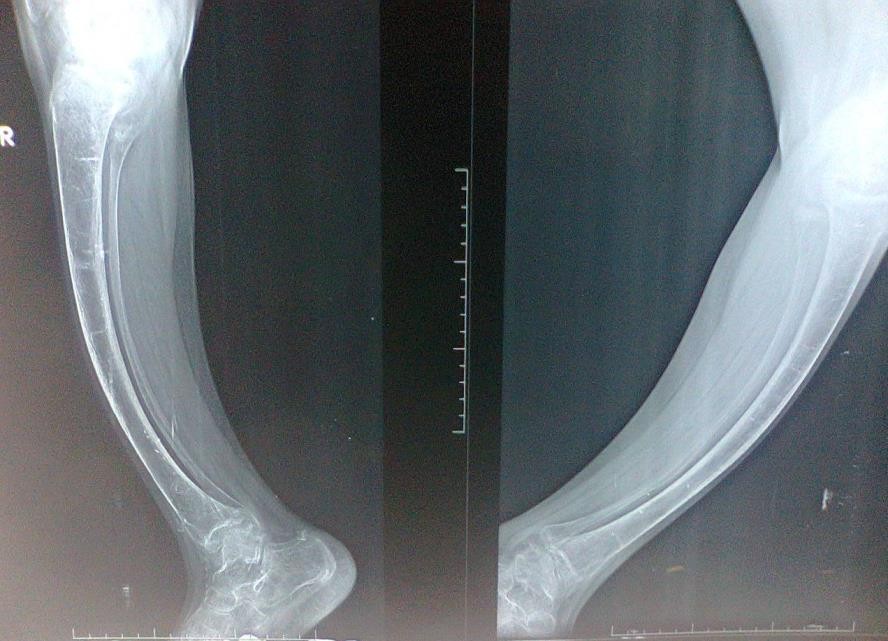 |
Figure-2C: Radiographs showedmore severe bowing
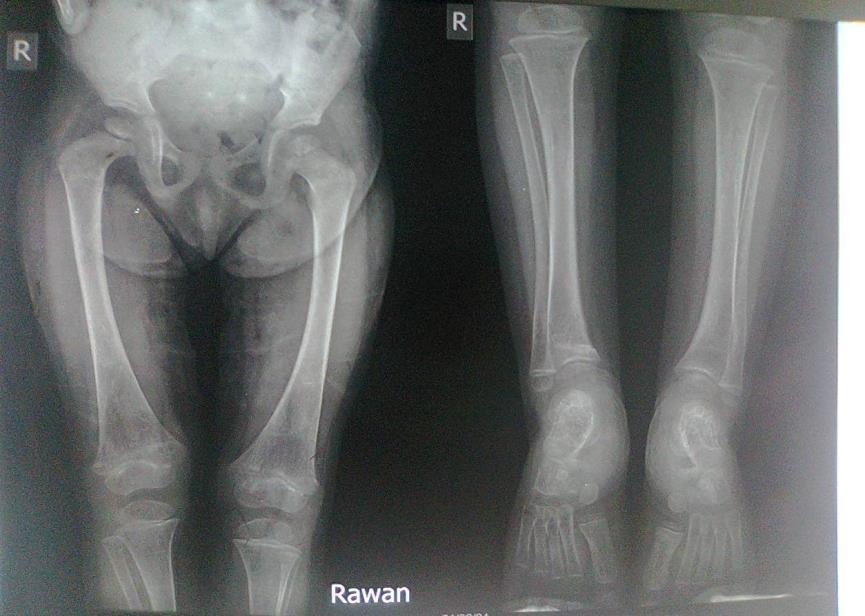
Figure-3A:lower limb radiograph showing progressive bowing
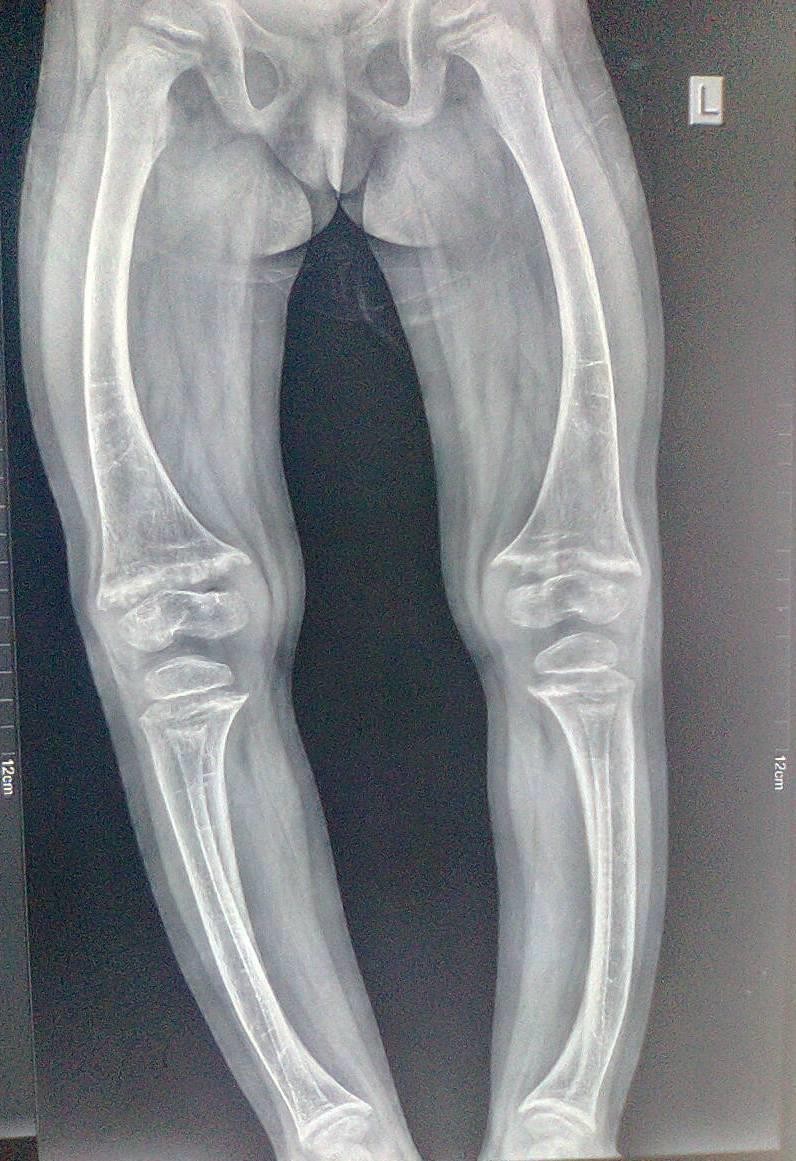 |
Figure-3B:lower limb radiograph showing progressive bowing
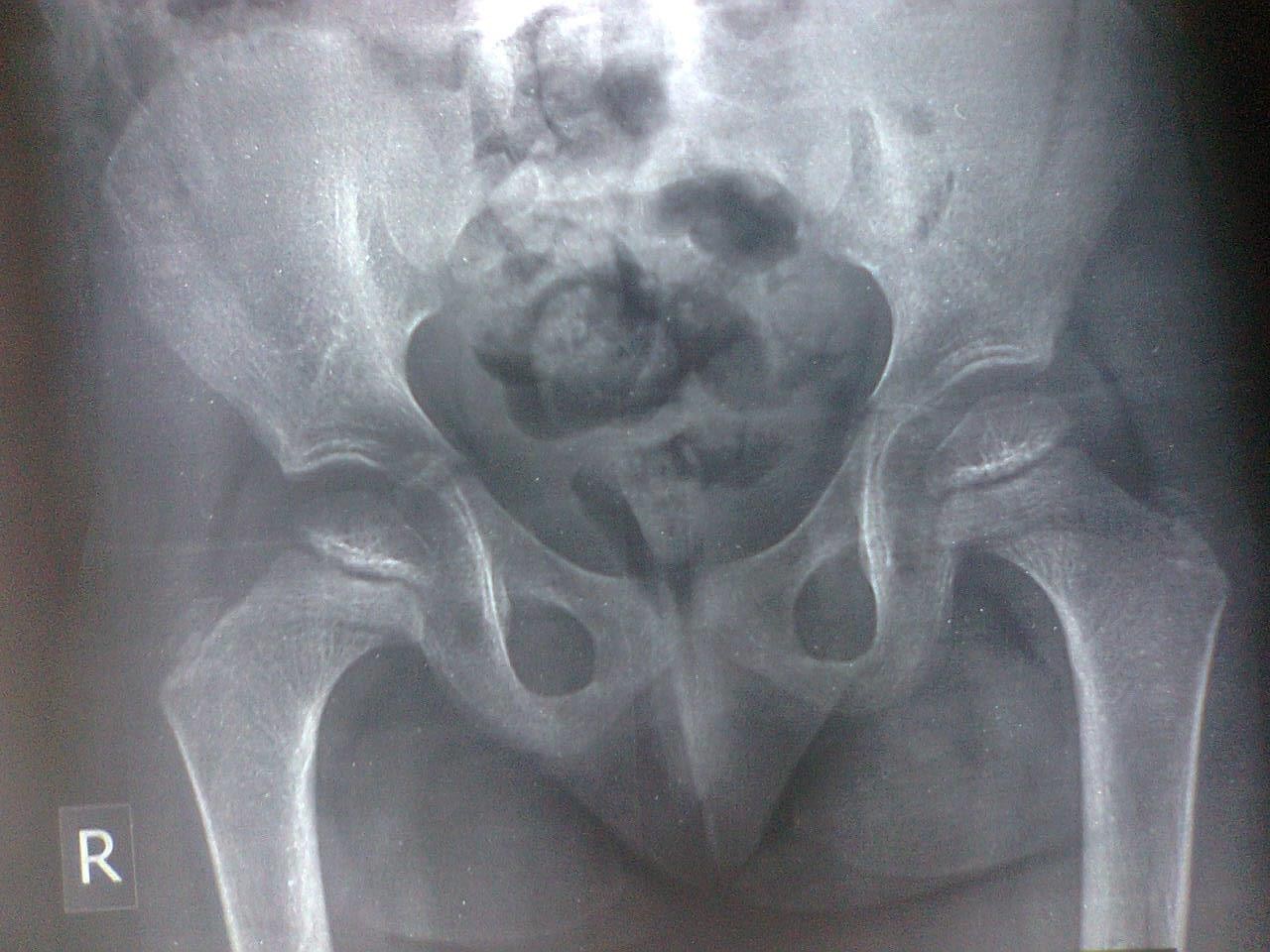
Figure-4A:Radiograph of the pelvis showingmild protrusio acetabula
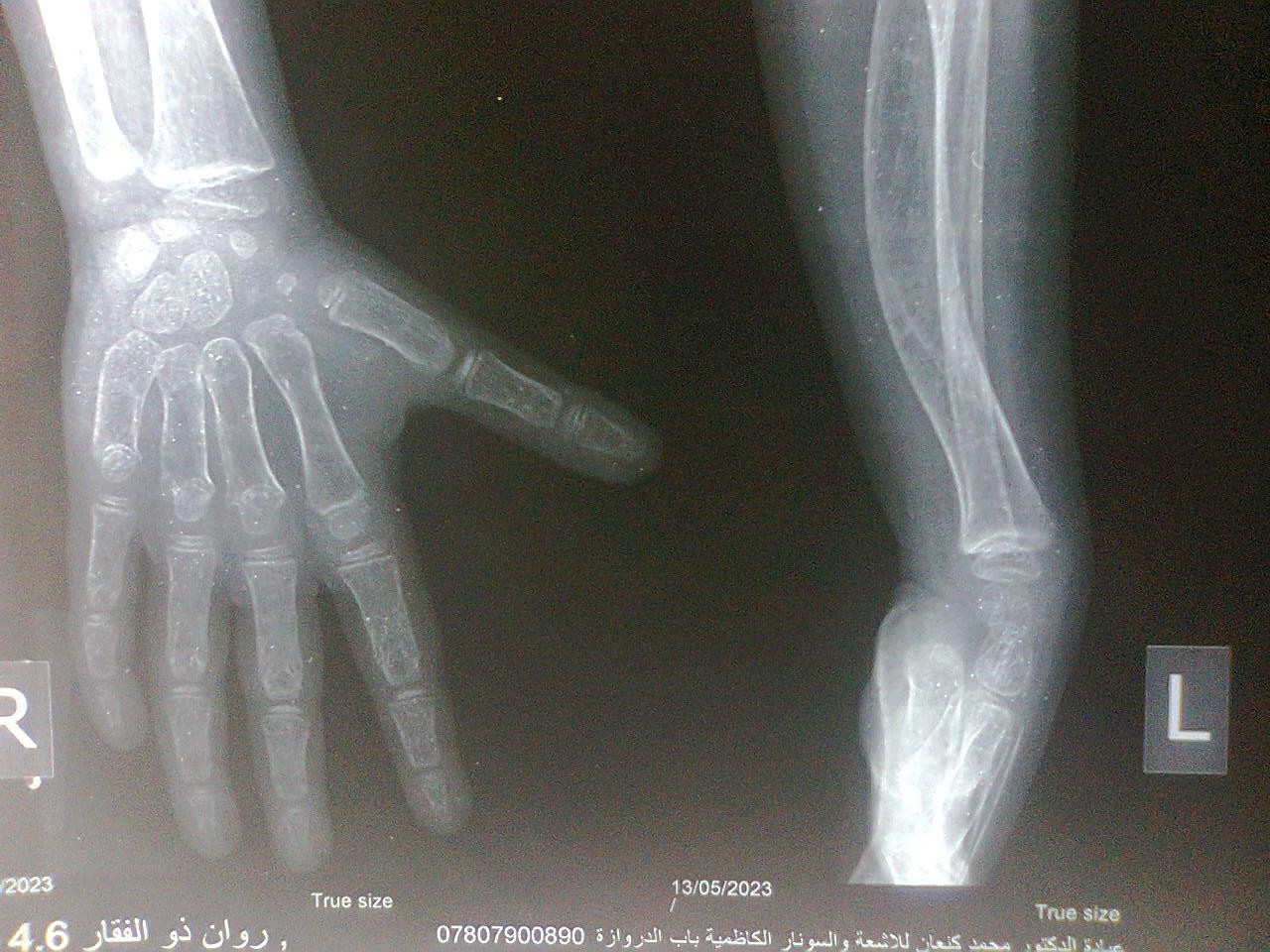 |
Figure-4B:Radiograph of the wrist showingno rachitic changes
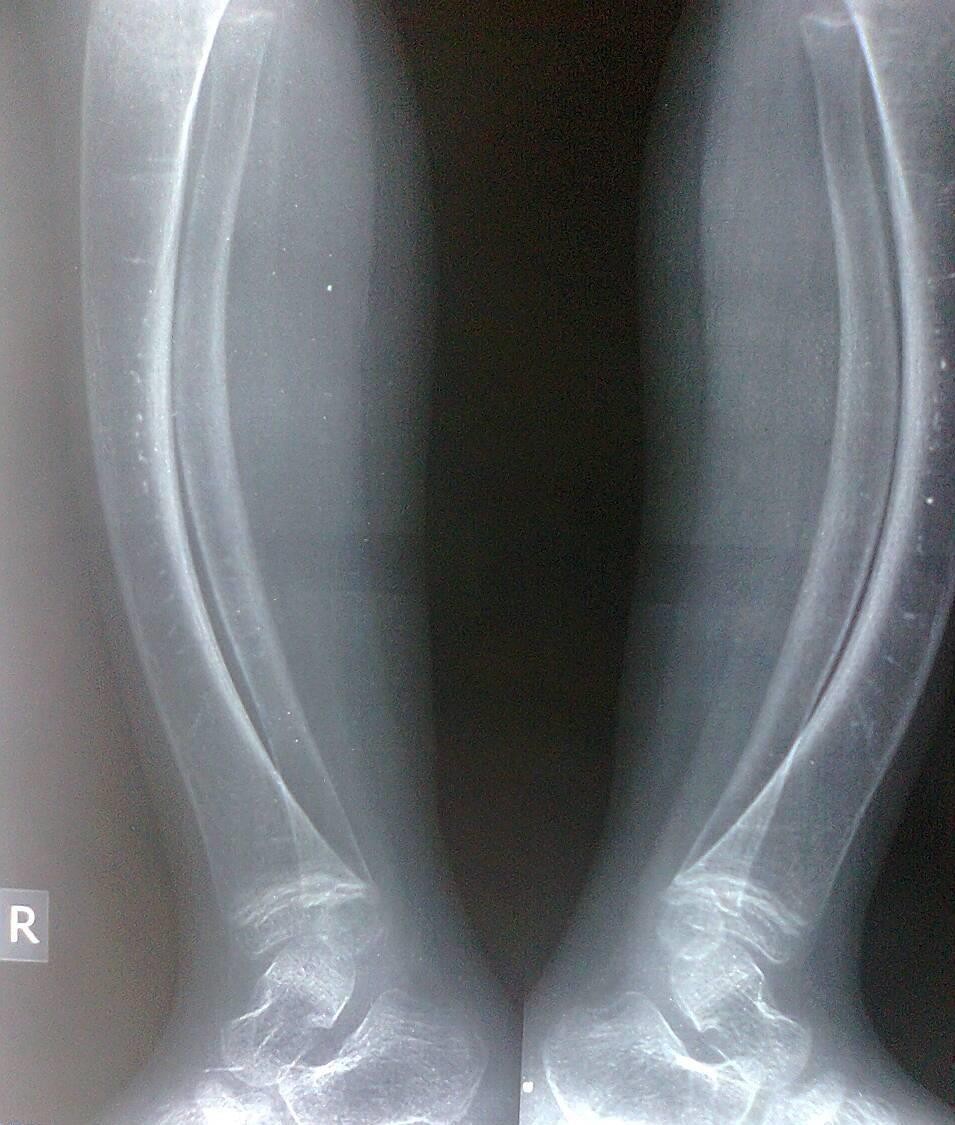
Figure-4C:Radiograph of the lower limbs showing bowing,cortical thinning with scanty spongiosa
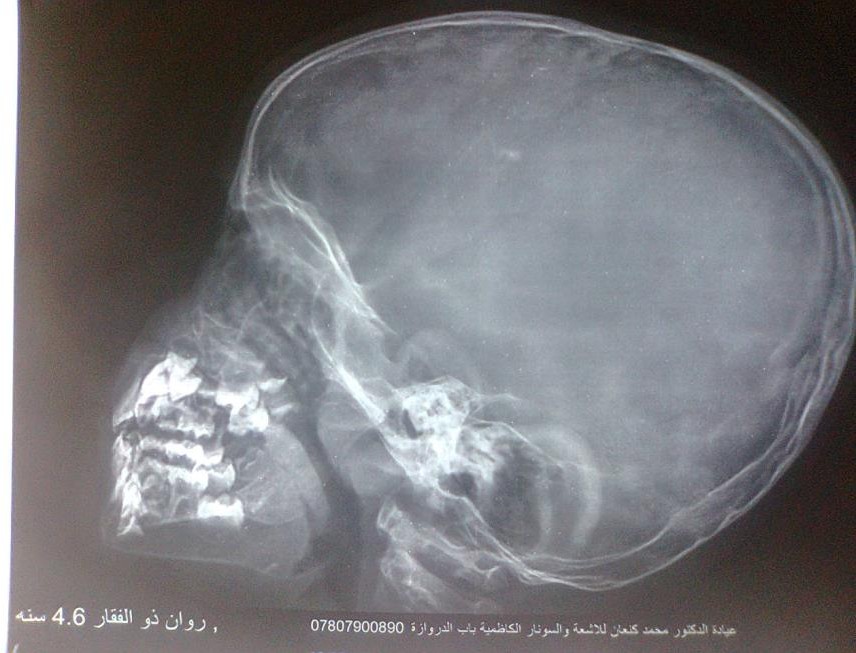 |
Figure-4D: Lateral skull radiographs showed platybasia
Discussion
The main aim of treatment of Ekman-Lobstein syndrome is strengthening the fragile bones and preventing the occurrence of fracture. Therefore, biphosphonates including pamidronate, alendronate, and zoledronic acid have been increasingly used, and intravenous bisphosphonates are generally
treatment for the disorder[1, 2].
Pamidronate is usuallygiven as a slowintravenous infusion over about three hours, and is generally given every three to six months and usually needed for the life.
As early as 1998, Francis H Glorieux (Figure-5) from Canada and his research team reported that intravenous pamidronate can reduce bone pain, prevent fractures, reshape existing fractures, and reduce long-bonefractures. However, the occurrence of bone pain,hypocalcemia, nausea, and dizziness has observed in some patients.
The study reported by Glorieux and his researchteam included thirtypatients with severe form of Ekman-Lobstein syndrome aged 3 to 16 years. They were treated with intravenous pamidronate every four to six months for 1.3 to 5 years.
Treatment was associated with increased bone density and decreased mean incidence of fractures. Treatment was also associated with decreased bone resorption and a decline in serum alkaline phosphatase and reduction in urinary excretion of calcium and type I collagen N-telopeptide [3].

Figure-5: Francis H Glorieux
In 2006, Linda A DiMeglio and Munro Peacock from the United States reported a study which included 18 patients older than three years with Ekman-Lobstein syndrome. Nine children were treated with oral alendronate 1 mg/kg/daily, and nine were treated with intravenous pamidronate 3mg/kg every four months. The study showed that both treatments were equally effective especially in less severe disease [5].
In 2017, Otilia Marginean from Romania and her research group reported the treatment of 9 children with various types of Ekman-Lobstein syndrome with pamidronate 1 mg/kg every3 months. Bone pain was lessened after the first treatment session with increased activity and mobility. Treatment was associated with improved bone density in association with lowering of alkaline phosphatase [6].
In 2013, Nick Bishop from the United Kingdom and his research group reported a one-year placebo-controlled study which included 143 children aged 4-15 years with Ekman-Lobstein syndrome. 94 patients were treated with oral risedronate, a third-generation bisphosphonates in a dose of 2·5 or 5 mg daily and 49 patients served as the placebo group. Treatment was associated with increased bone density and decreased the risk of fractures. Risedronate was generally well tolerated [7].
A 2-yearstudy was conductedby Lv et al in 2018, and included 136 children and adolescents aged 2 to 16 years with Ekman-Lobstein syndrome. 90 patients received weekly oral alendronate 70 mg, and 46 patients received yearly infusion of zoledronic acid for 2 years. The study showed that the yearly 5 mg infusion of zoledronic acid and weekly oral alendronate were associated with similar effects in increasing bone mineral density and decreasing bone resorption in children and adolescents with Ekman-Lobstein syndrome. However, zoledronic infusion was superior to alendronate in decreasing the clinical fracture rate [8].
Conclusion
This paper documented the occurrence of another type of Ekman-Lobstein syndrome in Iraq without blue sclerae, in addition to type 1 (David Owen Sillence and colleagues’ classification) which we have previously reported. The current expert opinion suggests that bisphosphonates have been shown to be beneficial in improving the condition and therefore should be considered as the treatment of choice.
Acknowledgement
The author has the copy right of all the sketchesincluded in this paper.
Conflict of interest:None.
References
- Al-Mosawi AJ. Ekman-Lobstein syndrome. 1st ed, Saarbrücken. (2018). LAP Lambert Academic Publishing, (ISBN: 978-613- 9-87306-7).
View at Publisher | View at Google Scholar - Al-Mosawi AJ. (2019). Ekman-Lobstein syndrome: Clinical, radiologic features, and the therapeutic challenge. Global Journal of Orthopedics Research (ISSN: 2687-816X) 13:2(1):1- 4.
View at Publisher | View at Google Scholar - Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R. (1998). Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med, 339
View at Publisher | View at Google Scholar - Sillence DO, Senn A, Danks DM. (1979). Genetic heterogeneity in osteogenesis imperfecta. J Med Genet, 16(2):101-116.
View at Publisher | View at Google Scholar - DiMeglio LA, Peacock M. (2006). Two-year clinical trial of oral alendronate versus intravenous pamidronate in children with osteogenesis imperfecta. J Bone Miner Res, 21(1): 132-140.
View at Publisher | View at Google Scholar - Marginean O, Tamasanu RC, Mang N, Mozos I, Brad GF. (2017). Therapy with pamidronate in children with osteogenesis imperfecta. Drug Des Devel Ther, 28:11:2507-2515.
View at Publisher | View at Google Scholar - Bishop N, Adami S, Ahmed SF, Antón J, Arundel P, Burren CP, Devogelaer JP, Hangartner T, Hosszú E, Lane JM, Lorenc R, Mäkitie O, Munns CF, Paredes A, Pavlov H, Plotkin H, Raggio CL, Reyes ML, Schoenau E, Semler O, Sillence DO, Steiner RD. (2013). Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial. Lancet, Oct 26:382(9902):1424-1432.
View at Publisher | View at Google Scholar - Lv F, Liu Y, Xu X, Song Y, Li L, Jiang Y, Wang O, Xia W, Xing X. (2018). Zoledronic acid versus alendronate in the treatment of children with osteogenesis imperfecta: A 2-year clinical study. Endocr Pract, 24(2):179-188.
View at Publisher | View at Google Scholar
