

Case Report | DOI: https://doi.org/10.31579/2834-8788/31
A Report of Claudin-19 Mutation Causing Nephrocalcinosis and End-Stage Kidney Disease from Iran
1Professor of Nephrology, Firoozgar Research Development Center, School of Medicine, Iran University of Medical Sciences, Tehran, Iran
2Assistant Professor of Nephrology, Firoozgar Research Development Center, School of Medicine, Iran University of Medical Sciences, Tehran, Iran
*Corresponding Author: Saghar Chehrazi, Assistant Professor of Nephrology, Firoozgar Research Development Center, School of Medicine, Iran University of Medical Sciences, Tehran, Iran.
Citation: Shokoufeh Savaj, Saghar Chehrazi, (2025), A Report of Claudin-19 Mutation Causing Nephrocalcinosis and End-Stage Kidney Disease from Iran, Journal of Heart and Vasculature, 4(3); DOI:10.31579/2834-8788/31
Copyright: © 2025, Saghar Chehrazi. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 31 May 2025 | Accepted: 18 June 2025 | Published: 27 June 2025
Keywords: kidney stone; end stage kidney disease; hypomagnesemia; claudin-19; nephrocalcinosis
Abstract
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) is an autosomal recessive disorder that affect children and young adults. Mutation in gene that coding the tight junction proteins Claudin-16 and Claudin-19(CLDN19) is responsible of this rare disorder. Hypomagnesemia, hypercalciuria, kidney failure and visual impairment (in CLDN 19 gene mutation) are the most common presentations of FHHNC. Here we present a 31-year-old woman with end-stage kidney disease (ESKD) on routine hemodialysis for the past eight years and was referred to Firoozgar nephrology clinic for kidney transplantation. Her past medical history included recurrent kidney stones. Although FHHNC is a rare disease, genetic evaluation recommended in patients with ESKD and concomitant nephrocalcinosis.
IJKD 2024; 18:236-8 www.ijkd.org
Introduction
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) is an autosomal recessive condition that affect children and young adults. It is characterized by the excessive loss of magnesium and calcium through the kidneys, nephrocalcinosis, and kidney failure.1,2 Nephrolithiasis and recurrent urinary tract infections, are the most presentation of FHHNC.3,4,5 Approximately half of these patients need renal replacement therapy during their second or third decade of life.6 Extra-renal involvements of this syndrome include seizure, muscular spasms and eye involvement such as macular coloboma, nystagmus, retinopathy, and visual loss .5 Mutation in gene that codes the tight junction Claudin-16 and Claudin-19 (CLDN19) proteins is responsible of this rare disorder.1,7 To the best of our knowledge we report the first report of the Claudin -19 mutation with end-stage kidney disease (ESKD) and recurrent kidney stones in Iran.
Case Report
A 31-year-old woman with ESKD was referred to Firoozgar nephrology clinic for a kidney transplant workup. She was on Hemodialysis for the past eight years and had a history of recurrent kidney stones that required surgical intervention and extracorporeal shock wave lithotripsy. The patient ’s brother had recurrent kidney stones and never followed kidney evaluation. There was no history of bone fractures, or visual or hearing loss. On physical examination, she had short stature, pigmented skin, and nystagmus with no skeletal deformity. Demographic data consisted of the height of 147 Cm, weight of 44 kg, body mass index of 20.4 kg/m2, and blood pressure of 130/75 mm/Hg. Lab tests have shown in Table 1. Ophthalmologic study showed a bilateral macular scar, nystagmus, and cataract. An echocardiogram revealed an ejection fraction of 55%, mild left ventricular diastolic dysfunction, mild to moderate tricuspid regurgitation, and normal pulmonary arterial pressure. Ultrasound study showed bilateral small size kidneys, an eight mm stone in the inferior calyx of the right kidney, and a simple cyst in the superior pole of the left kidney. Whole A Report of Claudin-19 Mutation Causing Nephrocalcinosis—Savaj et al exome sequencing (WES) was performed to rule out of hyperoxaluria. Whole exome sequencing and variant data analysis revealed a homozygote variant defined as c.364G > A (p. Gly122Arg) in exon 2 of CLDN19 gene (NM_148960.3) which compatible with FHNCC. AGXT, GRHPR, HOGA1 genes for hyperoxaluria showed no mutation.
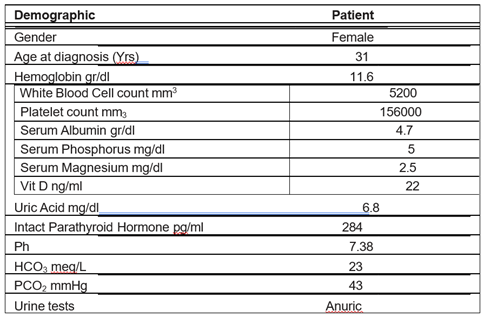
Table 1: Demographic Characteristics and Laboratory Results
Discussion
Most of the filtered Magnesium is reabsorbed by a passive paracellular mechanism in the thick ascending limb of Henle (TAL).8 Claudins are tight junction proteins, in thick ascending loop of Henle (TAL) and distal convoluted tubules (DCT) that regulate the paracellular cation transport.9,10 Impaired functions of either claudin-16 or claudin-19 result in renal magnesium and calcium wasting.11 FHHNC is an autosomal recessive disorder resulting from mutations in the CLDN16 or CLDN19 genes.12 It presents with hypercalciuria, magnesuria, hypomagnesemia and nephrocalcinosis. Additional symptoms include nephrolithiasis, urinary tract infections, polyuria, polydipsia, hypocitraturia, muscle spasms and convulsions.3,6 Patients with cludin19 mutation also have eye involvement as same as severe myopia, nystagmus, and chorioretinitis.5 Our patient had nephrolithiasis, ESKD, mild ocular abnormalities and, a missense mutation with a homozygote variant defined as c.364G > A (p.Gly122Arg) in exon 2 of CLDN19 gene (NM_148960.3). Amar et al, described three children in one family with a homozygous missense mutation in CLDN19 (c.241C > T, p.Arg 81Cys) with severe ocular involvement and retinal changes. 13 Another presentation is a report of a 33-year-old Chinese woman with a history of recurrent acute pyelonephritis who developed ESKD after three years with a homozygous mutation (c.346C > G, p. Leu116Val) in the 115G-L-W117 motif of CLDN 16 gene.12 In one study, 31 patients with four pathogenic mutations (p. G122R, p.I41T, p.G75C, and p.G75S) of the CLDN19 gene were presented. They had the age range of 6.8-48, and ocular manifestation, such as nystagmus, myopia, and macular coloboma were observed in 87% of the patients.14 Malakoutian et al. reported a pathogenic variant of a missense mutation (c.338G > A: p.C113Y) in exon 2 of the CLDN-16 gene in a 35 years-old-man with ESKD on routine hemodialysis and his two siblings. 15 To the best of our knowledge, our patient is the first reported case of the CLDN19 gene mutation in Iran. We should consider FHNCC as a possible differential diagnosis of recurrent nephrolithiasis with hypomagnesemia in patients with nephrocalcinosis.
Statement of Ethics
The study was approved by Iran University of Medical Sciences ethics committee IR.IUMS.FMD. REC. 1402.329. The patient’s written informed consent was secured including the permission to publish anonymously.
Conflict of Interest
The authors declare no conflict of interest.
References
- Manz F, Scharer K, JankaP, Lombeck J. Renal magnesium wasting, incomplete tubular acidosis, hypercalciur and nephrocalcinosis in siblings.Eur J Pediatr1978;128:67-79
View at Publisher | View at Google Scholar - Weber S, Hoffmann K, Jeck N, et al. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis maps to chromosome 3q27 disassociated with mutations in the PCLN-1 gene. Eur J Hum Genet 2000; 8:414-22.
View at Publisher | View at Google Scholar - Rodr ́ıguez-Soriano J, Vallo A, Garc ́ıa-Fuentes M (1987). Hypomagnesaemia of hereditary renal origin. Pediatr Nephrol 1: 465–472.
View at Publisher | View at Google Scholar - Weber S, Schneider L, Peters M, Misselwitz J, Ronnefarth G, et al. (2001) Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 12: 1872–1881.
View at Publisher | View at Google Scholar - Benigno V, Canonica CS, Bettinelli A, von Vigier RO, Truttmann AC, et al. (2000) Hypomagnesaemia- hypercalciuria-nephrocalcinosis: A report of nine cases
View at Publisher | View at Google Scholar - A Report of Claudin-19 Mutation Causing Nephrocalcinosis—Savaj et al and a review. Nephrol Dial Transplant 15: 605–610.
View at Publisher | View at Google Scholar - Rodriguez Soriano J, Vallo A. Pathophysiology of the renal acidification defect present in the syndrome of familial hypomagnesaemia hypercalciuria. Pediatr Nephrol 1994; 8:431-5.
View at Publisher | View at Google Scholar - Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez- Soriano J, McCredie D, Milford D, Sanjad S, Lifton
View at Publisher | View at Google Scholar - RP. Paracellin-1, a real tight junction protein required for paracellular Mg2+ resorption. Science. 1999; 285(5424):103–6.
View at Publisher | View at Google Scholar - Alexander RT, Hoenderop JG, Bindels RJ. Molecular determinants of magnesium homeostasis: insights from human disease. J Am Soc Nephrol 2008; 19: 1451–1458.
View at Publisher | View at Google Scholar - Hou J, Renigunta A, Konrad M, et al. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. Clin Invest 2008; 118:619-28.
View at Publisher | View at Google Scholar - Haisch L, Almeida JR, Abreu da Silva PR, Schlingmann KP, Konrad M. The role of tight junctions in paracellular ion transport in the renal tubule: lessons learned from a rare inherited tubular disorder. Am J Kidney Dis 2011; 57:32030.
View at Publisher | View at Google Scholar - Hou J, Renigunta A, Gomes AS, et al. Good enough, claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci USA 2009; 106:15350-5.
View at Publisher | View at Google Scholar - Jingru Lu, Xiangzhong Zhao, Alessandro Paarden, Yanhua Lang, Irene Botello, Leping Shao, Familial hypomagnesaemia, Hypercalciuria and Nephrocalcinosis associated with a novel mutation of the highly conserved leucine residue 116 of Claudin 16 in a Chinese patient with a delayed diagnosis: a case report, Affiliations expand
View at Publisher | View at Google Scholar - Amar Al-Shibli, Martin Konrad, Waleed Altay, Omar Al Masri, Lihad Al-Gazali, Ibrahim Al Attrach Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC): report of three cases with a novel mutation in CLDN19 gene Saudi J Kidney Dis Transpl. 2013 Mar; 24(2):338-44.
View at Publisher | View at Google Scholar - Félix Claverie-Martín 1, Víctor García-Nieto, Cesar Loris, Gema Ariceta, Inmaculada Nadal, Laura Espinosa, Ángeles Fernández-Maseda, Montserrat Antón-Gamero, Africa Avila, Álvaro Madrid, Hilaria González-Acosta, Elizabeth Córdoba-Lanus, Fernando Santos, Marta
View at Publisher | View at Google Scholar - Gil-Calvo, Mar Espino, Elena García-Martinez, Ana Sanchez, Rafael Muley; RenalTube Group. Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis 2013; 8(1): e53151.
View at Publisher | View at Google Scholar - Tahereh Malakoutian, Bahareh Madadi , Siamak Saber, A Novel Mutation in CLDN16 Gene Causing Familial Hypomagnesemia, Hypercalciuria, Nephrocalcinosis in An Iranian Family,Iran j kidney disease 2022 May;16(3):209-213.
View at Publisher | View at Google Scholar - Saghar Chehrazi, Assistant Professor of Nephrology, Firoozgar Research Development Center, School of Medicine, Iran University of Medical Sciences, Tehran, Iran
View at Publisher | View at Google Scholar
