

Research Article | DOI: https://doi.org/10.31579/2835-7957/026
An Emergent Clade of Sars-Cov-2 Linked to Returned Travellers from Iran
- John-Sebastian Eden 1,2*
- Rebecca Rockett 1 1,3,4
- Ian Carter 3
- Hossinur Rahman3 3
- Joep de Ligt 5
- James Hadfield 6
- Matthew Storey 5
- Xiaoyun Ren 5
- Rachel Tulloch 1,2
- Kerri Basile 3*
- Jessica Wells 3
- Roy Byun 7
- Nicky Gilroy 3
- Matthew V O’Sullivan 3,4
- Vitali Sintchenko 1,2,3
- Sharon C Chen 1,3,4
- Susan Maddocks 3
- Tania C Sorrell 1,2,3
- Edward C Holmes 1,3
- Dominic E Dwyer 1,3,4
- Jen Kok 3,4
1 The University of Sydney, Marie Bashir Institute for Infectious Diseases and Biosecurity, School of Life and Environmental Sciences & School of Medical Sciences, NSW 2006, Australia.
2 Westmead Institute for Medical Research, Centre for Virus Research & Centre for Infectious Diseases and Microbiology, Westmead, NSW 2145, Australia.
3 Centre for Infectious Diseases and Microbiology Laboratory Services, NSW Health Pathology - Institute of Clinical Pathology and Medical Research, NSW 2145, Australia.
4 Centre for Infectious Diseases and Microbiology – Public Health, Westmead Hospital, Westmead NSW 2145, Australia
5 Institute of Environmental Science and Research, Porirua 5240, New Zealand.
6 Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA.
7 NSW Ministry of Health, North Sydney, NSW 2059, Australia.
8 The members of the 2019-nCoV Study Group are listed at the end of the article.
*Corresponding Author: John-Sebastian Eden, Kerri Basile, 2. Westmead Institute for Medical Research, Centre for Virus Research & Centre for Infectious Diseases and Microbiology, Westmead, NSW 2145, Australia, 3. Centre for Infectious Diseases and Microbiology Laboratory Servic
Citation: Jale Moradi, Mohsen Moghoofei, Amir H. Alvandi, Ramin Abiri, Ozlem Yagız (2023), An Emergent Clade of Sars-Cov-2 Linked to Returned Travellers from Iran, Clinical Reviews and Case Reports, 2(3); DOI:10.31579/2835-7957/026
Copyright: © 2023, Jale Moradi and Mohsen Moghoofei. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 03 May 2023 | Accepted: 15 May 2023 | Published: 22 May 2023
Keywords: covid-19; sars-cov-2; genome sequencing; phylogenetics
Abstract
The SARS-CoV-2 epidemic has rapidly spread outside China with major outbreaks occurring in Italy, South Korea and Iran. Phylogenetic analyses of whole genome sequencing data identified a distinct SARS-CoV-2 clade linked to travellers returning from Iran to Australia and New Zealand. This study highlights potential viral diversity driving the epidemic in Iran, and underscores the power of rapid genome sequencing and public data sharing to improve the detection and management of emerging infectious diseases.
Main Text
From a public health perspective, the real-time whole genome sequencing (WGS) ofemerging viruses enables the informed development and design of molecular diagnostic methods, and tracingpatterns of spreadacross multiple epidemiological scales (i.e., genomicepidemiology). However, WGS capacities and data sharing policies vary in different countries and jurisdictions, leading to potential sampling bias due to delayed or underrepresented sequencing data from some areas with substantial SARS-CoV-2 activity. Herein, we show that the genomic analysesof SARS-CoV-2 strainsfrom Australian returnedtravellers with COVID-19 disease may provide important insights into viral diversity present in regions currently lacking genomic data.
Sars-Cov-2 Emergenceand Dissemination
Inlate December 2019, a clusterof cases of pneumonia of unknown aetiology in Wuhan city,Hubei province, China was reported by health authorities [1]. A novel betacoronavirus, designated SARS-CoV-2, was identified as the causative agent [2] of the disease now known as COVID-19, with substantial human-to-human transmission [3]. To contain agrowing epidemic, Chinese authorities implemented strict quarantine measures in Wuhan and surrounding areas in Hubei province. Significant delays in the global spreadof the virus were achieved, but despite these measures, cases were exportedto other countries. As of 9 March 2020, these numbered more than 100 countries, on all continents except Antarctica; the total number of confirmed infections exceeded 110,000 and there were nearly 4,000 deaths [4]. Although the vast majority of cases have occurred in China, major outbreakshave also been reported in Italy, South Korea and Iran [5]. Importantly, there is widespread local transmission in multiple countries outside China following independent importations of infection from visitorsand returned travellers.
Whole Genome Sequencing of Sars-Cov-2 Cases in Australia and New Zealand
In New South Wales (NSW), Australia, WGS for SARS-CoV-2 was developed based on an existing amplicon-based Illumina sequencing approach[6]. Viral extractswere prepared fromrespiratory tract samples where SARS-CoV-2 was detected by RT-PCR using World Health Organization recommended primers and probes targeting the E and RdRp genes, and then reverse transcribed using SSIV VILO cDNA master mix. The viral cDNA was used as input for multiple overlapping PCR reactions (~2.5kb each) spanning the viral genome usingPlatinum SuperFi master mix (primers provided in Supplementary Table S1). Amplicons were pooled equally, purified and quantified. Nextera XT libraries were prepared and sequencing was performed with multiplexing on an Illumina iSeq (300 cycle flow cell). InNew Zealand, the ARTIC networkprotocol was used for WGS [7]. In short, 400bp tiling amplicons designed with Primal Scheme[8] were used to amplify viral cDNA prepared with SuperScript III. A sequence library was then constructed using the Oxford NanoPoreligation sequencing kit and sequenced on a R9.4.1 MinION flow-cell. Near-complete viral genomeswere then assembled de novo in Geneious Prime 2020.0.5 or through reference mapping with RAMPART V1.0.6 [9] using the ARTIC network nCoV-2019 novel coronavirus bioinformatics protocol[10]. In total,13 SARS-CoV-2 genomeswere sequenced from cases in NSW diagnosed between 24 Januaryand 3 March 2020, as well as a singlegenome from the first patientin Auckland, New Zealand sampledon 27 February 2020 (Table1).
| GISAID ID | Virus name | Location | Collection date | Travelhistory |
| EPI_ISL_408976 | 408976/Australia/Sydney-2/2020-01-22 | Sydney,Australia | 22-Jan-20 | China |
| EPI_ISL_407893 | 407893/Australia/NSW01/2020-01-24 | Sydney,Australia | 24-Jan-20 | China |
| EPI_ISL_408977 | 408977/Australia/Sydney-3/2020-01-25 | Sydney,Australia | 25-Jan-20 | China |
| EPI_ISL_413490 | 413490/New_Zealand/01/2020-02-27 | Auckland, New Zealand | 27-Feb-20 | Iran |
| EPI_ISL_412975 | 412975/Australia/NSW05/2020-02-28 | Sydney,Australia | 28-Feb-20 | Iran |
| EPI_ISL_413594 | 413594/Australia/NSW08/2020-02-28 | Sydney,Australia | 28-Feb-20 | SE Asia |
| EPI_ISL_413595 | 413595/Australia/NSW09/2020-02-28 | Sydney,Australia | 28-Feb-20 | SE Asia |
| EPI_ISL_413213 | 413213/Australia/NSW06/2020-02-29 | Sydney,Australia | 29-Feb-20 | Iran |
| EPI_ISL_413214 | 413214/Australia/NSW07/2020-02-29 | Sydney,Australia | 29-Feb-20 | None |
| EPI_ISL_413596 | 413596/Australia/NSW10/2020-02-28 | Sydney,Australia | 01-Mar-20 | SE Asia |
| EPI_ISL_413597 | 413597/AUS/NSW11/2020-03-02 | Sydney,Australia | 02-Mar-20 | Iran |
| EPI_ISL_413600 | 413600/AUS/NSW14/2020-03-03 | Sydney,Australia | 03-Mar-20 | None |
| EPI_ISL_413598 | 413598/AUS/NSW12/2020-03-04 | Sydney,Australia | 04-Mar-20 | Iran |
| EPI_ISL_413599 | 413599/AUS/NSW13/2020-03-04 | Sydney,Australia | 04-Mar-20 | Iran |
Table 1: SARS-CoV-2 genomes sequenced in this study
Australian and New Zealand sequences were aligned to global reference strains sourced from GISAID with MAFFT [11] and then compared phylogenetically using a maximum likelihood approach[12].
A Distinct Clade of Sars-Cov-2 Identified in Travellers Returned from Iran
The Australian strains of SARS-CoV-2 were dispersed across the global SARS-CoV-2 phylogeny (Figure 1A). The first four cases of COVID-19 diseasein NSW occurred between 24 and 26 January 2020, and these were closely related (with 1-2 SNPs difference) to the prototype strain MN908947/SARS-CoV-2/Wuhan-Hu-1, which is the dominant variant circulating in Wuhan. As the four patientsidentified in this period had recently returnedfrom China, this region was the likely source of infection. From 1 February 2020, travel to Australia from mainland China was restricted to returning Australian residents and their children, who were placed in home quarantine for 14 days. Despite the intensive testing of such returning travellers, no further cases of COVID-19 were detected in NSW until 28 February 2020, when SARS-COV-2 was detected in an individual returning from Iran (NSW05). A close contact of this individual also tested positive (NSW14) providing the first evidence of local transmission within NSW. This was followed by further Iran travel-linked cases in NSW (NSW06,NSW11, NSW12, NSW13)and New Zealand (NZ01). Of note, the genomes of all patients with a history of travel to Iran were part of a monophyletic group defined by three nucleotide substitutions (G1397A, T28688C & G29742T) in the SARS-CoV-2 genome relative to the Wuhan prototype strain (Figure 1B). G1397A and T28688C both occur in coding regions with G1397A producing a non- synonymous change (V378I) in the ORF1ab encoded non-structural protein 2 region. G29742T occurs in the 3’ UTR. In addition to the Australian and New Zealand strains, this clade also included a traveller who had returnedto Canada from Iran (BC_37_0-2), providing further evidence of its likely link to the Iranian epidemic. Indeed, a search of all currently available GISAID sequences and metadata revealed no other complete genome sequences from patientswith documented historyof travel to or residence in Iran (as of 9 March 2020). A search of partial sequences identified two SARS-CoV-2 sequences which originated in Iran (413553/IRN/Tehran15AW/2020-02-28 and 413554/IRN/Tehran9BE/2020-02-23) spanning a 363 nt region of the viral nucleoprotein (N). Although short in length, these two sequences covered one of the informative SNPs defining this clade - T28688C, and both Iranian strains matchedthe sequences from patients with travel historiesto Iran and grouped by phylogenetic analysis(Supplementary Figures S1 & S2).
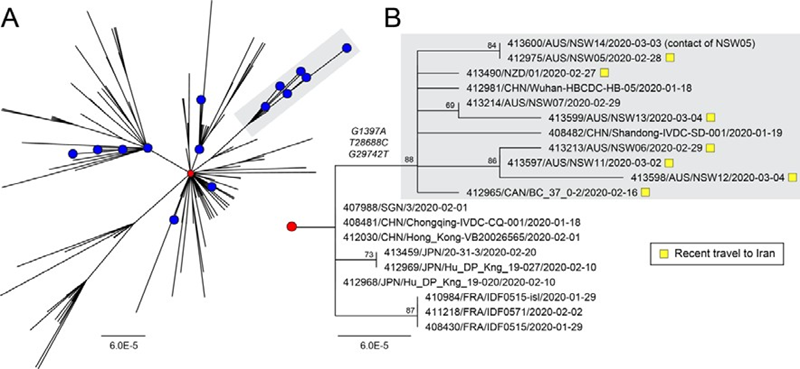
Figure 1 - Phylogenetic analysis of SARS-CoV-2 genome sequences highlighting a clade of importedcases from Iran. (A) Global diversityof circulating SARS-CoV-2strains including Australian sequences (blue circles, n=19). The prototype strain Wuhan-Hu-1 is shown as a red circle.An emergent clade containing casesimported from Iran is highlighted with grey shading.(B) Sub-tree showingthe informative branch containing importedIranian cases (highlighted with yellow squares) and defined by substitutions at positions G1397A, T28688C, G29742T. Node support is provided as bootstrap values of 100 replicates. For both panels A & B, the scales are proportional to the number of substitutions per site.
Discussion
Technological advancements and the wide-spread adoption of WGS in pathogen genomics have transformed public health and infectious disease outbreak responses [13]. Previously, disease investigations often relied on the targeted sequencing of a small locus to identify genotypes and infer patterns of spread along with epidemiological data. As seen with the recent West African Ebola [14] and Zika virus epidemics [15], rapid WGS significantlyincreases resolution of diagnosis and surveillance thereby strengthening links between clinical and epidemiological data [16]. This advance improves our understanding of pathogen origins and spread that ultimately lead to stronger and more timely intervention and control measures [17]. Following the first release of the SARS-CoV-2 genome [18], public health and researchlaboratories worldwide have rapidly shared sequences on public data repositories such as GISAID [19] (n = 236 genomes as of 9 March 2020) that have been used to provide near real-time snapshots of global diversity through public analytic and visualization tools [20].While all known cases linked to Iran are contained in this clade, it is important to note the presence of two Chinese strains sampled during mid-January 2020 from Hubei and Shandong provinces. It is expected that further Chinese strains would be identified within this clade, and across the entire diversity of SARS-CoV-2 as this is where the outbreak started, including for the outbreak in Iran itself. However, while we cannot completely discount that the cases in Australia and New Zealand came from other sources including China, our phylogenetic analyses, as well as epidemiological (recent travel to Iran) andclinical data (date of symptom onset), provide evidence that this clade of SARS-CoV-2 is linked to the Iranianepidemic, from where genomic data is currently lacking. Importantly, the seemingly multiple importations of very closely related viruses from Iran into Australia suggests that this diversityreflects the early stages of SARS-CoV-2 transmission within Iran.
Acknowledgments
The members of the nCoV-2019 Study Group include Linda Donovan, Shanil Kumar, Tyna Tran, Danny Ko, Christine Ngo, Tharshini Sivaruban, Verlaine Timms, Connie Lam, Mailie Gall, Karen-Ann Gray, Rosemarie Sadsad and Alicia Arnott. The authors acknowledge the Sydney Informatics Hub and the use of the University of Sydney’s high performance computingcluster, Artemis, and all the laboratories that have referredSARS-CoV-2 samples to the Centre for Infectious Diseases and Microbiology Laboratory Services, NSW HealthPathology - Instituteof Clinical Pathologyand Medical Research,We stmead Hospital. We would finally like to thank all the authors who have kindly shared genome data on GISAID,and have includeda table (Supplementary Table S2) outlining the authors and institutes involved. Data including the sequences in this study are available for download from
Author Contributions
Study concept and design by JSE, ECH & JK. Sample processing and testing by IC & HR. Sequencing and analysis by JSE, RR, JDL, JH, MS, XR, RT & ECH. Study coordination by NG, MVOS, VS, SCC, SM, TCS, DED & JK. JSE wrote the first manuscript draft with editingfrom ECH, JDL, RR, TCS, VS &JK. The final manuscript was approved by all authors.
Conflict Of Interest
None declared.
Funding
This study was supported by the Prevention Research Support Program funded by the New South Wales Ministry of Health and the NHMRC Centre of Research Excellence in Emerging Infectious Diseases (GNT1102962).
References
- Wuhan Municipal Health and Health Commission’s briefing on the current pneumonia epidemic situation in our city (2019).
View at Publisher | View at Google Scholar - Wu F, Zhao S, Yu B, Chen Y-M, Wang W, et al. (2020). A new coronavirus associated with human respiratory disease in China. Nature.
View at Publisher | View at Google Scholar - Lu R, Zhao X, Li J, Niu P, Yang B, et al. (2019). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet;395(10224):565–574
View at Publisher | View at Google Scholar - Dong E, Du H, Gardner L. (2020). An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis.
View at Publisher | View at Google Scholar - World Health Organisation Coronavirus Situation Report - 8th March (2020).
View at Publisher | View at Google Scholar - Di Giallonardo F, Kok J, Fernandez M, Carter I, Geoghegan JL, et al. (2018). Evolution of Human Respiratory Syncytial Virus (RSV) over Multiple Seasons in New South Wales, Australia. Viruses.
View at Publisher | View at Google Scholar - nCoV-2019 sequencing protocol, Quick J.
View at Publisher | View at Google Scholar - Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, et al. (2019). An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol.
View at Publisher | View at Google Scholar - ARTIC Network RAMPART [Internet]. [cited 2020 Mar 10]. Available from: https://github.com/artic-network/rampart
View at Publisher | View at Google Scholar - ARTIC Network Bioinformatics SOP [Internet]. [cited 2020 Mar 10]. Available from: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html
View at Publisher | View at Google Scholar - Katoh K, Misawa K, Kuma K-I, Miyata T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 15;30(14):3059–3066.
View at Publisher | View at Google Scholar - Guindon S, Gascuel O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. Oct;52(5):696–704
View at Publisher | View at Google Scholar - Popovich KJ, Snitkin ES. (2017). Whole Genome Sequencing-Implications for Infection Prevention and Outbreak Investigations. Curr Infect Dis Rep;19(4):15
View at Publisher | View at Google Scholar - Dudas G, Carvalho LM, Bedford T, Tatem AJ, Baele G, et al. (2017). Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature.;544(7650):309–315.
View at Publisher | View at Google Scholar - Grubaugh ND, Faria NR, Andersen KG, Pybus OG. (2018). Genomic Insights into Zika Virus Emergence and Spread. Cell.;172(6):1160–1162
View at Publisher | View at Google Scholar - Grenfell BT, Pybus OG, Gog JR, Wood JLN, Daly JM, et al. (2004). Unifying the epidemiological and evolutionary dynamics of pathogens. Science.303(5656):327–332
View at Publisher | View at Google Scholar - Grubaugh ND, Ladner JT, Lemey P, Pybus OG, Rambaut A, et al. (2019). Tracking virus outbreaks in the twenty-first century. Nature Microbiology. 4(1):10-19
View at Publisher | View at Google Scholar - Virological.org - Novel 2019 coronavirus genome. Holmes EC et al. [Internet]. [cited 2020 Mar 9]. Available from: http://virological.org/t/novel-2019-coronavirus-genome/319
View at Publisher | View at Google Scholar - Shu Y, McCauley J. (2017). GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. ;22(13)
View at Publisher | View at Google Scholar - Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, et al. (2018). Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. ;34(23):4121–4123
View at Publisher | View at Google Scholar
